
ГОСТ 30404-94 (ИСО 157-75)
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ТОПЛИВО ТВЕРДОЕ МИНЕРАЛЬНОЕ
МЕТОДЫ ОПРЕДЕЛЕНИЯ ФОРМ СЕРЫ
Издание официальное
Б3 9-
МЕЖГОСУДАРСТВЕННЫЙ СОВЕТ
ПО СТАНДАРТИЗАЦИИ, МЕТРОЛОГИИ И СЕРТИФИКАЦИИ
Минск
ГОСТ 30404-94
Предисловие
1 РАЗРАБОТАН МТК 179 «Уголь и продукты его переработки», институтом горючих ископаемых (ИГИ)
ВНЕСЕН Госстандартом России
2 ПРИНЯТ Межгосударственным Советом по стандартизации, метрологии и сертификации (протокол № 6—94 от 21 октября 1994 г.)
За принятие проголосовали:
Наименование государства | Наименование национального органа по стандартизации |
Азербайджанская Республика Республика Армения Республика Белоруссия Республика Грузия Республика Казахстан Киргизская Республика Республика Молдова Российская Федерация Республика Узбекистан Украина | Аз госстандарт Арм госстандарт Бе л стандарт Груэстандарт Госстандарт Республики Казахстан Киргизстандарт Молдовастандарт Госстандарт России Узгосстандарт Госстандарт Украины |
3 Настоящий стандарт представляет собой полный аутентичный текст ИСО 157—75 «Уголь каменный. Методы определения форм серы» и содержит дополнительные требования, отражающие потребности экономики страны
4 ВВЕДЕН ВПЕРВЫЕ
5 Постановлением Комитета Российской Федерации по стандартизации, метрологии и сертификации от 9 июлй 1996 г. № 444 межгосударственный стандарт ГОСТ 30404—94 (ИСО 157—75) введен в действие непосредственно в качестве государственного стандарта Российской Федерации с I июля 1997 г.
© ИПК Издательство стандартов, 1997
Настоящий стандарт не может быть полностью или частично воспроизведен, тиражирован и распространен в качестве официального издания на территории Российской Федерации без разрешения Госстандарта России
Содержание
1 Назначение и область применения
2 Нормативные ссылки
3 Сущность метода
3.1 Общие положения
3.2 Сульфатная сера
3.3 Пиритная сера
3.4 Сульфидная сера
3.5 Органическая сера
3.6 Пересчеты на другие состояния топлива
4 Подготовка проб
5 Определение сульфатной серы гравиметрическим методом ... 4
6 Определение пиритной серы методом окисления
7 Определение пиритной серы методом восстановления.....
8 Определение сульфидной серы
9 Вычисление массовой доли органической серы
10 Протокол определения
Приложение А Пояснения к терминам, применяемым
в стандарте
ГОСТ 30404-94 (ИСО 157-75)
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ТОПЛИВО ТВЕРДОЕ МИНЕРАЛЬНОЕ
Метода определения форм серы
Solid fuel. Determination of forms of sulphur
Дата введения 1997—07—01
1 НАЗНАЧЕНИЕ И ОБЛАСТЬ ПРИМЕНЕНИЯ
Настоящий стандарт распространяется на бурые и каменные угли, антрацит, лигниты, горючие сланцы, кокс, торф, твердые продукты обогащения и переработки (в дальнейшем — топливо) и устанавливает методы определения форм соединений серы: сульфатной, пиритной, сульфидной и способ расчета органической серы.
Пояснения к терминам, применяемым в настоящем стандарте, приведены в приложении А.
Дополнительные требования, отражающие потребности экономики страны, выделены курсивом.
2 НОРМАТИВНЫЕ ССЫЛКИ
В настоящем стандарте использованы ссылки на следующие стандарты:
ГОСТ 61—75 Кислота уксусная. Технические условия
ГОСТ 1277—75 Серебро азотнокислое
ГОСТ 1770— 74 Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Технические условия
ГОСТ 3118—77 Кислота соляная. Технические условия
ГОСТ 3760—79 Аммиак водный. Технические условия
ГОСТ 4108—72 Барий хлористый. Технические условия
ГОСТ 4145—74 Калий сернокислый. Технические условия
ГОСТ 4159—79 Йод. Технические условия
ГОСТ 4220—75 Калий двухромовокислый. Технические условия
ГОСТ 4232—74 Калий йодистый. Технические условия
Издание официальное
ГОСТ 4461—77 Кислота азотная. Технические условия
ГОСТ 4517—87 Реактивы. Методы приготовления вспомогательных реактивов и растворов, применяемых при анализе
ГОСТ 4919.1—77 Реактивы и особо чистые вещества. Методы приготовления растворов индикаторов
ГОСТ 5824— 79 Кадмий уксуснокислый 2-водный. Технические условия
ГОСТ 8050—85 Двуокись углерода газообразная и жидкая. Технические условия
ГОСТ 9147—80 Посуда и оборудование лабораторные фарфоровые. Технические условия
ГОСТ 10163—76 Крахмал растворимый. Технические условия
ГОСТ 10742—71 Угли бурые, каменные, антрацит, горючие сланцы и угольные брикеты. Методы отбора и подготовки проб для лабораторных испытаний
ГОСТ 10929—76 Водорода перекись. Технические условия
ГОСТ 11303—75 Торф. Метод приготовления аналитических проб
ГОСТ 18300—87 Спирт этиловый ректификованный технический. Технические условия
ГОСТ 23083—78 Кокс каменноугольный, пековый и термоантрацит. Методы отбора и подготовки проб для испытаний
ГОСТ 25336—82 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 27313—951 Топливо твердое. Обозначения аналитических показателей и формулы пересчета результатов анализа для различных состояний топлива.
ГОСТ 27314—91 Топливо твердое минеральное. Методы определения влаги
ГОСТ 29169—91 Посуда лабораторная стеклянная. Пипетки с одной отметкой
ГОСТ 29252—91 Посуда лабораторная стеклянная. Бюретки без времени ожидания.
3 СУЩНОСТЬ МЕТОДА
3.1 Общие положения
Сущность методов определения форм серы зависит от того, в виде каких соединений сера присутствует в топливе.
Обычно сера в твердом топливе содержится в виде неорганических сульфатов, железных колчеданов (FeS2), сульфидов металлов и органических соединений серы.
В соответствии с этим известны следующие формы соединений серы в топливе: сульфатная, пиритная, сульфидная и органическая.
3.2 Сульфатная сера
Сульфатную серу определяют, экстрагируя содержащиеся в минеральной части топлива сульфаты разбавленной соляной кислотой. В экстрактах серу определяют в виде сульфат-ионов гравиметрически (раздел 4).
3.3 Пиритная сера
3.3.1 Пиритная сера не растворяется в разбавленной соляной кислоте, но количественно растворяете# в разбавленной азотной кислоте в условиях, установленных настоящим стандартом.
Пиритную серу определяют косвенным методом окисления по количеству железа, связанного с серой в FeS2 (серных колчеданах) (раздел 5).
3.3.2 Другой метод заключается в восстановлении пиритной серы водородом до сероводорода, улавливании выделяющегося сероводорода раствором ацетата кадмия и йодометрическом титровании образовавшегося сульфида кадмия с учетом содержания в топливе сульфидной серы. Пробу топлива предварительно доизмельчают для раскрытия зерен пирита (раздел 6).
3.4 Сульфидная сера
При обработке топлива кислотами сульфиды металлов разлагаются с выделением сероводорода.
Метод определения сульфидной серы заключается в обработке топлива соляной кислотой, улавливании выделяющегося сероводорода раствором ацетата кадмия и йодометрическом титровании образовавшегося сульфида кадмия (раздел 7).
3.5 Органическая сера
Органическую серу рассчитывают по разности между общей серой и суммой разновидностей серы: сульфатной, пиритной и сульфидной в процентах (раздел 10),
3.6 Пересчеты на другие состояния топлива
Результаты определения рассчитывают на аналитическое состояние топлива. Пересчет результатов на другие состояния топлива производят по ГОСТ 27313.
4 ПОДГОТОВКА ПРОБ
Для определения форм соединений серы используют аналитическую пробу топлива крупностью зерен менее 0,2 мм.
Аналитическую пробу топлива готовят по ГОСТ 10742, ГОСТ 11303 и ГОСТ 23083 в зависимости от вида топлива. Пробу выдерживают в тонком слое минимально необходимое время для достижения приблизительного равновесия между влагой пробы и влажностью лабораторного помещения.
Перед выполнением определения воздушно-сухую пробу перемешивают не менее 1 мин.
Для определения пиритной серы методом восстановления (раздел 6) пробу доизмельнают для раскрытия зерен пирита до крупности не более 0,02 мм (6.4.1).
Одновременно определяют массовые доли влаги топлива по ГОСТ 27314 и общей серы по ГОСТ 8606.
5 ОПРЕДЕЛЕНИЕ СУЛЬФАТНОЙ СЕРЫ ГРАВИМЕТРИЧЕСКИМ МЕТОДОМ
5.1 Реактивы
Все реактивы должны быть квалификации ч.д.а.; для анализа применяют только дистиллированную воду.
5.1.1 Соляная кислота по ГОСТ 3118, плотностью 1,18 г/см3.
5.1.2 Соляная кислота, раствор. К 420 см3 соляной кислоты (5.1.1) добавляют воды до 1 дм3.
5.1.3 Соляная кислота, раствор. К 42 см3 соляной кислоты (5.1.1) добавляют воды до 1 дм3.
5.1.4 Барий хлористый (ВаСЬ2Н2О) по ГОСТ 4108, 8,5 %-ный (85 г/дм3) раствор. 100 г двуводного хлорида бария растворяют в воде и разбавляют до 1 дм3 раствора. Раствор фильтруют через плотный фильтр или прокладку из фильтровальной бумаги, дважды промытые кислотой (5.1.3).
5.1.5 Аммония гидроксид по ГОСТ 3760, раствор плотностью около 0,88 г/см3.
5.1.6 Бромная вода по ГОСТ 4517. Готовят насыщенный раствор брома в воде.
5.1.7 Стандартный раствор сульфата: 0,6000 г сульфата калия (АГ25О4 по ГОСТ 4145) растворяют в воде в мерной колбе вместимостью 1 дм3 и доливают водой до метки.
10 см3 стандартного раствора сульфата соответствуют 0,0080 г сульфата бария (BaSO4).
5.1.8 Метиловый оранжевый, индикатор, 0,1 %-ный раствор по ГОСТ 4919.1. 0,1 г метилового оранжевого (диметил-амино-азобен-золсульфокислота) растворяют в 100 см3 воды при нагревании, при необходимости охлажденный раствор фильтруют.
5.1.9 Метиловый красный, индикатор, 0,1 %-ный раствор в 60 %-ном этиловом спирте по ГОСТ 4919.1.
0,1 г метилового красного растворяют в 60 см3 этилового спирта и добавляют воды до 100 см3.
5.1.10 Водорода пероксид по ГОСТ 10929, 3 %-ный раствор.
5.1.11 Серебро азотнокислое (AgNOJ по ГОСТ 1277, 3 %-ный раствор, подкисленный азотной кислотой.
5.2 Аппаратура
Используют стеклянную посуду по ГОСТ 1770, ГОСТ 9147, ГОСТ 20292 и ГОСТ 25336.
Весы аналитические с погрешностью взвешивания не более 0,1 мг. Допускается применять аналитические весы с погрешностью взвешивания не более 0,2 мг.
5.2.1 Печь муфельная электрическая с терморегулятором для поддержания температуры (8ОО±25) *С.
Плита толщиной 6 мм из керамики (или другого подходящего изоляционного материала), которая легко вставляется в печь.
5.2.2 Штифтовый холодильник (рисунок 1).

Рисунок 1 —• Штифтовый холодильник
5.2.3 Обратный холодильник типа ХШ-1 или 3 по ГОСТ 25336.
5.3 Проведение анализа
5.3.1 Навеску топлива массой около 5 г помещают в коническую колбу вместимостью 250 см3, добавляют 50 см3 раствора соляной кислоты (5.1.2), внутрь горла колбы вставляют штифтовый (5.2.2) или обратный (5.2.3) холодильник. Допускается проводить экстракцию топлива в химическом стакане без холодильника; в этом случае объем раствора соляной кислоты увеличивают до 100 см*.
Суспензию кипятят в течение 30 мин и фильтруют через фильтр средней плотности, предварительно дважды промытый кислотой (5.1.3).
5.3.2 Остаток на фильтре промывают 6 раз раствором соляной кислоты (5.1.3) порциями приблизительно по 20 см3.
В фильтрат (200—250 см3) приливают 1 см3 бромной воды (5.1.6) или 10 см* пероксида водорода (5.1.10) и кипятят в течение 5 мин для превращения всего растворенного непиритного железа в трехвалентное.
5.3.3 Осаждают ионы железа, медленно, по каплям приливая к кипящему раствору раствор аммиака (5.1.5) до окончания образования осадка и еще 5 см3 в избыток. Раствор быстро фильтруют в стакан через неплотный фильтр.
Осадок на фильтре (А) промывают горячей водой до отсутствия ионов хлора (проба с азотнокислым серебром, 5.1.11).
Осадок А сохраняют для определения непиритного железа (6.4.1.6).
5.3.4 Фильтрат (5.3.2) выпаривают до объема 150—200 см*. Добавляют 10,0 см3 стандартного раствора сульфатов (5.1.7). Приливают 2—3 капли индикатора метилового оранжевого (5.1.8) или метилового красного (5.1.9) и затем осторожно соляную кислоту (5.1.1) до изменения цвета раствора и еще 1 см3 в избыток. Объем раствора должен составлять от 150 до 250 см3.
Содержимое стакана кипятят 5 мин для удаления диоксида углерода, затем уменьшают нагрев и продолжают нагревать, не доводя его до кипения.
Добавляют 10 см3 холодного раствора хлористого бария (5.1.4) по каплям из пипетки в течение 20 с так, чтобы хлорид бария попадал в центр горячего раствора при постоянном перемешивании.
В течение дальнейших 30 мин раствор выдерживают при температуре, близкой к температуре кипения без перемешивания (примечание 1 к 5.3.5).
5.3.5 Фильтруют, используя один из двух методов (примечание 2):
1 Под действием силы тяжести через беззольный плотный фильтр диаметром 100—125 мм, дважды промытый кислотой, вложенный в химическую воронку типа В-75—110(170) по ГОСТ 25336.
2 Под действием силы тяжести через фильтровальную прокладку, приготовленную из беззольной, дважды промытой кислотой фильтровальной бумаги.
Осадок на фильтре промывают горячей водой до полного удаления ионов хлора. Отсутствие ионов хлора проверяют по реакции с азотнокислым серебром (5.1.11).
Влажный фильтр с осадком помещают в предварительно прокаленный и взвешенный фарфоровый, керамический или платиновый тигель, слегка уплотняют и осторожно высушивают и обугливают над горелкой, в сушильном шкафу или на электрической плитке, не допуская воспламенения, затем тигель прокаливают в муфельной печи при температуре (800^25) *С в течение 15 мин. Допускается помещать тигель с влажным фильтром с осадком на керамическую плиту и вносить в нагретую до (8ОО±25) °C муфельную печь на 15 мин (примечание 3).
Тигель вынимают из муфельной печи, охлаждают сначала на воздухе, а затем в эксикаторе до комнатной температуры и взвешивают.
Примечания
1 Время выстаивания осадка. Известно, что полнота образования осадка сульфата бария достигается при выдерживании раствора в течение 30 мин в рекомендуемых условиях: солянокислая среда (концентрация соляной кислоты в растворе составляет приблизительно 0,05 н.) и значительный избыток ионов бария. При таких условиях осадок сульфата бария полностью выпадает в течение 30 мин выдерживания раствора при температуре, близкой к температуре кипения.
2 Фильтрование осадка. Для быстрого фильтрования используют метод 1. или 2.
Метод 1. Круглый фильтр должен быть тщательно подогнан к воронке.
Метод 2. Для того, чтобы приготовить фильтровальную прокладку, нарезанные кусочки из фильтровальной бумаги, дважды промытой кислотой, площадью около 1 см2 помещают в колбу с дистиллированной водой и встряхивают до образования бумажной массы.
В химическую воронку диаметром 75 мм помещают фарфоровую коническую насадку для фильтрования диаметром 25 мм, стебель воронки закрывают пальцем и наливают дистиллированную воду так, чтобы насадка и стебель были заполнены водой. На коническую насадку помещают бумажную массу и формуют прокладку толщиной 5 мм с помощью стеклянной палочки с плоским концом. Сливают избыток воды, удаляя палец от стебля воронки, и слегка уплотняют прокладку по краям, используя ту же палочку. После промывки фильтровальная прокладка готова к применению.
После окончания фильтрования раствора и промывания осадка прокладку с осадком переносят в тигель, стенки воронки протирают двумя половинками беззольного фильтра, который затем помещают в тигель и прокаливают вместе с осадком.
3 Прокаливание. В предложенных условиях прокаливание влажного фильтра с осадком происходит быстро; использование толстой изоляционной плиты предотвращает механические потери.
5.4 Контрольный опыт
Контрольный опыт проводят в тех же условиях, но без навески топлива. Перед добавлением индикатора (5.1.8 или 5.1.9) в раствор добавляют 10 см3 стандартного раствора сульфата калия (5.1.7).
Массу сульфата бария, полученную в контрольном опыте, вычитают из массы, полученной при определении с навеской.
5.5 Обработка результатов
Массовую долю сульфатной серы в аналитической пробе в процентах вычисляют по формуле где т — масса навески, г;
0,1374 (тх-т£
т
тх — масса сульфата бария, полученная при определении, г;
т2 — масса сульфата бария, полученная в контрольном опыте, г; 0,1374 — коэффициент пересчета сульфата бария на серу.
Результаты (предпочтительно среднее значение двух определений) округляют до 0,1 %.
5.6 Точность метода
5.6.1 Сходимость
Результаты параллельных определений, выполненных в разное время в одной и той же лаборатории одним лаборантом при использовании одной и той же аппаратуры из одной и той же аналитической пробы, не должны превышать значений, указанных в таблице 1.
Таблица 1
Сульфатная сера | Максимально допускаемые расхождения между результатами . (рассчитанными для одинакового содержания влаги) |
в одной лаборатории (сходимость) | в разных лабораториях (воспроизводимость) |
0,02 % абс | 0,03 % абс |
Если расхождение между результатами двух определений превышает значения, указанные в таблице, то проводят третье определение и за результат принимают среднее арифметическое результатов двух наиболее близких результатов в пределах допускаемых расхождений.
Если результат третьего определения находится в пределах допускаемых расхождений по отношению к каждому из двух предыдущих определений, то за результат анализа принимают среднее арифметическое результатов трех определений.
5.6.2 Воспроизводимость
Средние значения результатов параллельных определений, выполненных в двух разных лабораториях на представительных порциях, взятых из одной и той же пробы после последней стадии ее подготовки, не должны превышать значений, указанных в таблице 1.
6 ОПРЕДЕЛЕНИЕ ПИРИТНОЙ СЕРЫ МЕТОДОМ ОКИСЛЕНИЯ
6.1 Общие положения
Метод основан на окислении пиритной серы азотной кислотой до получения растворимых сульфатов и определении ее титрованием по пиритному железу.
Определение пиритной серы проводят из одной навески, последовательно обрабатывая ее соляной и азотной кислотой (6.4.1), или из двух навесок, причем одну обрабатывают соляной, а вторую — азотной кислотами (6.4.2). В обоих случаях в солянокислый раствор переходит непиритное железо, а в азотнокислой при первом способе определения — только пиритное железо, а при втором ~ сумма пиритного и непиритного железа.
Железо в растворах определяют йодометрическим (6.4.3), комплекснометрическим (6.4.4) или бихроматным (6.4.5) титрованием.
Обработку всех видов топлива азотной кислотой проводят при комнатной температуре в течение 24 или 2 ч при непрерывном встряхивании. Экстракцию каменных углей и антрацитов азотной кислотой можно, кроме того, проводить при кипячении со штифтовым (5.2.2) или обратным (5.2.3) холодильником в течение 30 мин.
Топлива низких стадий метоморфизма (торф, лигниты, бурые угли) при кипячении с разбавленной азотной кислотой частично растворяются, причем окраска раствора не исчезает при нагревании с перекисью водорода и титрометрическое определение железа становится невозможным.
6.2 Реактивы
Все реактивы должны быть квалификации чд.а. Для анализа применяют только дистиллированную воду.
6.2.1 Азотная кислота по ГОСТ 4461, раствор. К 125 см3 азотной кислоты плотностью 1,42 г/см3 добавляют воды до 1 дм3.
6.2.2 Водорода перекись по ГОСТ 10929, раствор 300 г/дм3 (30 %-ный).
6.2.3 Соляная кислота по 5.1.2.
6.2.4 Смесь серной и фосфорной кислот. 150 см3 серной кислоты плотностью 1,84 и 150 см3 фосфорной кислоты плотностью 1,75 г/см3 осторожно смешивают. Эту смесь приливают в 600 см3 воды, охлаждают и доливают водой до 1 дм3.
6.2.5 Хлорид олова (II), раствор 50 г/дм3. 5 г безводного хлорида олова (II) или 6 г двуводной соли растворяют в 50 см3 соляной кислоты
(плотностью 1,18 г/см3) и этот раствор приливают в 50 см3 воды. Раствор готовят непосредственно перед употреблением.
6.2.6 Хлорид ртути (И), раствор 60 г/дм3. 6 г хлорида ртути (II) добавляют в 100 см3 воды и перемешивают смесь в течение 10 мин.
6.2.7 Аммония гидроксид по 5.1.5.
6.2.8 Калий двухромовокислый (бихромат) по ГОСТ 4220, раствор 0,0030 моль/дм3. 0,8780 г бихромата калия растворяют в воде и разбавляют до 1 дм3.
6.2.9 Дифениламиносульфонат натрия по ГОСТ 49191, индикатор, раствор 2 г/дм3. 0,2 г дифениламиносульфоната натрия растворяют в воде и разбавляют до 100 см3. Хранят в темной склянке.
6.2.10 Калий иодистый по ГОСТ 4232, раствор 200 г/дм3.
6.2.11 Натрий серноватистокислый (тиосульфат натрия) Na/Sfl?
■ 5Н2О, раствор 0,05моль/дм3. 12,410 г тиосульфата натрия растворяют в небольшом объеме воды в мерной колбе вместимостью 1 дм? и доливают водой до метки.
6.2.12 Крахмал растворимый, индикатор, по ГОСТ 10163, 1 %-ный раствор, приготовленный по ГОСТ 4517.
6.2.13 Кислота сульфосалициловая (C7H6O6S2H2O) по ГОСТ 4478, 10 %-ный раствор.
6.2.14 Соль динатриевая этилендиамин — N, N, N1, Н-тетрауксус-ной кислоты двуводная (du-Na-ЭДТА) по ГОСТ 10652, раствор 0,05 моль/дм3. Приготовление раствора из стандарт-титра или по ГОСТ 10398.
6.3 Аппаратура
Используют стеклянную посуду по ГОСТ 1770, ГОСТ 9147, ГОСТ 25336, ГОСТ 29169 и ГОСТ 29252.
Весы аналитические с погрешностью взвешивания не более 0,1 мг.
Допускается применять аналитические весы с погрешностью взвешивания не более 0,2 мг.
6.4 Проведение анализа
6.4.1 Определение пиритной серы из одной навески топлива.
6.4.1.1 Навеску топлива массой 1 г обрабатывают разбавленной соляной кислотой по 5.3.1. Остаток на фильтре промывают 4—5 раз горячей водой.
6.4.1.2 Фильтр с остатком помешают в стакан или колбу вместимостью 250—300 см3, приливают 50—80 см3 азотной кислоты по 6.2.1, тщательно перемешивают и в соответствии с 6.1 оставляют стоять при комнатной температуре на 24 ч, или колбу с остатком закрывают пробкой и перемешивают на встряхивателе в течение 2 ч, или к колбе присоединяют штифтовый (5.2.2) или обратный (5.2.3) холодильник и кипятят содержимое в течение 30 мин.
6.4.1.3 Нерастворимый остаток фильтруют через фильтр средней плотности, предварительно дважды промытый азотной кислотой, и 7—8 раз промывают горячей водой. Фильтрат и промывные воды собирают в один стакан.
6.4.1.4 Если фильтрат окрашен, приливают 2—3 см3 перекиси водорода (6.2.2) и кипятят 5 мин для обесцвечивания раствора.
6.4.1.5 Для осаждения гидроксида железа (III) к кипящему раствору медленно приливают раствор аммиака (6.2.7) до небольшого избытка (до устойчивого запаха) и еще добавляют 5 см3. Раствор фильтруют через неплотный фильтр и осадок гидроксида железа (III) промывают горячей водой с несколькими каплями аммиака.
6.4.1.6 Для количественного перенесения осадка гидроксида железа (III) фильтр прокалывают и смывают осадок тонкой струей горячей воды в коническую колбу или стакан вместимостью 500 см3. На фильтр небольшими порциями приливают соляную кислоту (6.2.3) для удаления следов железа и окончательно промывают фильтр горячей водой. Для полного растворения гидроксида железа (III) содержимое колбы нагревают.
Можно также для количественного перенесения гидроксида железа (III) в колбу растворить осадок на фильтре горячей соляной кислотой (6.2.3), а затем промыть фильтр горячей водой. Далее железо (III) определяют по 6.4.3 или 6.4.4, или 6.4.5.
6.4.2 Определение пиритной серы из двух навесок топлива.
6.4.2.1 Одну навеску топлива массой 1—5 г обрабатывают раствором соляной кислоты по 5.3.1—5.3.3. Осадок А сохраняют для определения непиритного железа (6.4.1.6 и далее по 6.4.3, или 6.4.4, или 6.4.5).
6.4.2.2 Вторую навеску топлива обрабатывают раствором азотной кислоты по 6.4.1.2.
Обработку суспензии ведут по 6.4.1.3—6.4.1.6.
6.4.3 Йодометрическое титрование железа в растворе.
Раствор, полученный при растворении гидроксида железа в разбавленной соляной кислоте (6.4.1.5), нейтрализуют раствором гидроксида аммония (6.2.7), приливая его по каплям до появления осадка. Затем к раствору приливают 10 см3 соляной кислоты (6.2.3), 10 см3 раствора йодистого калия (6.2.10), накрывают колбу часовым стеклом и ставят в темное место на 5 мин. Выделившийся йод титруют тиосульфатом натрия (6.2.11) до перехода окраски в соломенно-желтую, затем приливают растворимый крахмал (6.2.12) и продолжают титрование тиосульфатом натрия до полного обесцвечивания раствора.
6.4.4 Комплексонометрическое титрование железа в растворе.
Раствор, полученный при растворении гидроксида железа в разбавленной соляной кислоте (6.4.1.5), нейтрализуют раствором гидроксида аммония (6.2.7), приливая его по каплям до появления осадка. Затем приливают при перемешивании по каплям раствор соляной кислоты (6.2.3) до pH 1,4—1,8 и добавляют 1—2 см3 раствора сульфосалициловой кислоты (6.2.13). Раствор нагревают до 55—60 'Сив горячем состоянии титруют раствором du-Na-ЭДТА (6.2.14) до перехода красно-фиолетовой окраски в зеленовато-желтую или до обесцвечивания раствора в зависимости от содержания железа.
Допускается комплексонометрическое титрование железа без предварительного выделения гидроксида железа в бесцветном фильтрате, полученном по 6.4.1.2—6.4.1.4.
6.4.5 Бихроматное титрование железа в растворе
Раствор, полученный при растворении гидроксида железа (III) в соляной кислоте, выпаривают до объема 20 см3. Раствор нагревают до кипения и добавляют по каплям из пипетки или из капельной воронки раствор хлорида олова (II) (6.2.5) при перемешивании до исчезновения желтой окраски раствора. Добавляют 5 капель в избы* ток, охлаждают до комнатной температуры и быстро приливают 10 см3 раствора хлорида ртути (II) (6.2.6). Образуется осадок хлорида ртути (I). Добавляют 15 см3 смеси серной и фосфорной кислот (6.2.4), разбавляют водой до объема 150—200 см3, добавляют 5 капель индикатора (6.2.9) и титруют раствором бихромата калия (6.2.8). Около конечной точки титрования цвет раствора становится голубовато-зеленым, а в присутствии большого количества железа — зеленовато-голубым. По каплям добавляют бихромат калия, пока цвет раствора не станет фиолетово-голубым.
6.5 Контрольный опыт
Контрольный опыт проводят в тех же условиях, но без навески топлива.
6.6 Обработка результатов
6.6.1 При определении пиритной серы из одной навески с йодометрическим окончанием (6.4.1 и 6.4.3) массовую долю пиритной серы в аналитической пробе (S?) в процентах вычисляют по формуле
= ^0.064 ^
р ту 9
где V — объем раствора тиосульфата натрия, израсходованного на титрование пробы, см3;
V] — объем раствора тиосульфата натрия, израсходованного на титрование в контрольном опыте, см3;
М — концентрация раствора тиосульфата натрия, моль/дм3;
0,064 — количество пиритной серы (г), соответствующее 1 см3 раствора тиосульфата натрия с концентрацией 1 моль/дм3;
т3 — навеска топлива, г.
6.6.2 При определении пиритной серы из одной навески с комплексонометрическим окончанием (6.4.1 и 6.4.4) массовую долю пиритной серы в аналитической пробе S?, %, вычисляют по формуле
s. 0.064 ■
где V2 — объем раствора du-Na-ЭДТА, израсходованного на титрование пробы, см3;
V3 — объем раствора du-Na-ЭДТА, израсходованного на титрование в контрольном опыте, см3;
Mj — концентрация раствора du-Na-ЭДТА, моль/дм3;
0,064 — количество пиритной серы (г), соответствующее 1 см3 раствора du-Na-ЭДТА с концентрацией 1 моль/дм3.
6.6.3 При определении пиритной серы из двух навесок с бихро-матным окончанием (6.4.2 и 6.4.5) массовую долю пиритной серы в аналитической пробе S°, %, вычисляют по формуле
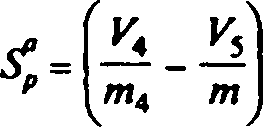
0,115,
где V4 — объем раствора бихромата калия, см3, израсходованного на титрование общего железа в навеске массой т4,
К5 — объем раствора бихромата калия, см3, израсходованного на титрование непиритного железа в навеске массой т.
6.6.4 Результаты (предпочтительно среднее арифметическое двух определений) округляют до 0,1 %.
6.7 Точность метода
Таблица 2
Пиритная сера. % | Максимально допекаемые расхождения между результатами (рассчитанными для одинакового содержания влаги) | |
в одной лаборатории (сходимость) | в разных лабораториях (воспроизводимость) | |
До 0,5 | 0,05 % абс | 0,10% абс |
0,5-1,5 | 0,07% абс | 0,15% абс |
Св. 1,5 | 5 % от результата | 10 % от результата |
6.7.1 Сходимость по 5.6.1.
6.7.2 Воспроизводимость — по 5.6.2.
7 ОПРЕДЕЛЕНИЕ ПИРИТНОЙ СЕРЫ МЕТОДОМ ВОССТАНОВЛЕНИЯ
7.1 Сущность метода — по 3.3.2.
7.2 Реактивы
7.2.1 Все реактивы должны быть квалификации ч.д.а.; для анализа применяют только дистиллированную воду.
7.2.2 Хром металлический, в порошке.
7.2.3 Цинк металлический, гранулированный.
7.2.4 Этанол по ГОСТ 18300, 96 %-ный.
7.2.5 Соляная кислота по 5.1.1.
7.2.6 Соляная кислота, раствор: к 525 см3 соляной кислоты (7.2.5) добавляют воды до 1 дм3.
7.2.7 Уксусная кислота по ГОСТ 61, ледяная, плотностью 1,05 г/см3.
7.2.8 Кадмий уксуснокислый, Cd (СН3СО0)2-2Н2О по ГОСТ 5824, раствор 43 г/дм3.
50 г двуводного уксуснокислого кадмия растворяют в воде в мерной колбе вместимостью 1 дм3, добавляют 10 см3 уксусной кислоты (7.2.7) и доливают водой до метки.
7.2.9 Натрий серноватистокислый (тиосульфат натрия) Na2S2Or5H2O, раствор 0,1 моль/дм3-25 г пятиводного тиосульфата натрия растворяют в свежекипяченой воде, добавляют 1 см3 хлороформа и разбавляют водой до 1 дм3. Концентрацию раствора устанавливают по раствору йодид-йодата калия (7.2.11).
7.2.10 Иод по ГОСТ 4159, раствор приблизительно 0,05 моль/дм3: 12,7 г йода растворяют в растворе, содержащем 25 г йодида калия в 35 см3 воды. После того, как весь йод растворится, разбавляют водой до 1 дм3. Концентрацию раствора устанавливают по раствору тиосульфата натрия (7.2.9).
7.2.11 Йодид-йодат калия, раствор 0,0052 моль/дм3 в расчете на йодат калия. 1,1146 г йодата калия и 10 г йодида калия растворяют в воде и разбавляют до 1 дм3.
7.2.12 Крахмал растворимый, индикатор по 6.2.12.
7.2.13 Диоксид углерода, газ по ГОСТ 8050 или азот, газ по ГОСТ 9293, в баллоне.
7.3 Аппаратура
Используют стеклянную посуду по ГОСТ 1770, ГОСТ 9147, ГОСТ 20292 и ГОСТ 25336.
Весы аналитические с погрешностью взвешивания не более 0,1 мг.
Допускается применять аналитические весы с погрешностью взвешивания не более 0,2 мг.
Прибор для определения пиритной серы, состоит из частей, указанных в 7.3.1—7.3.6 (рисунок 2).
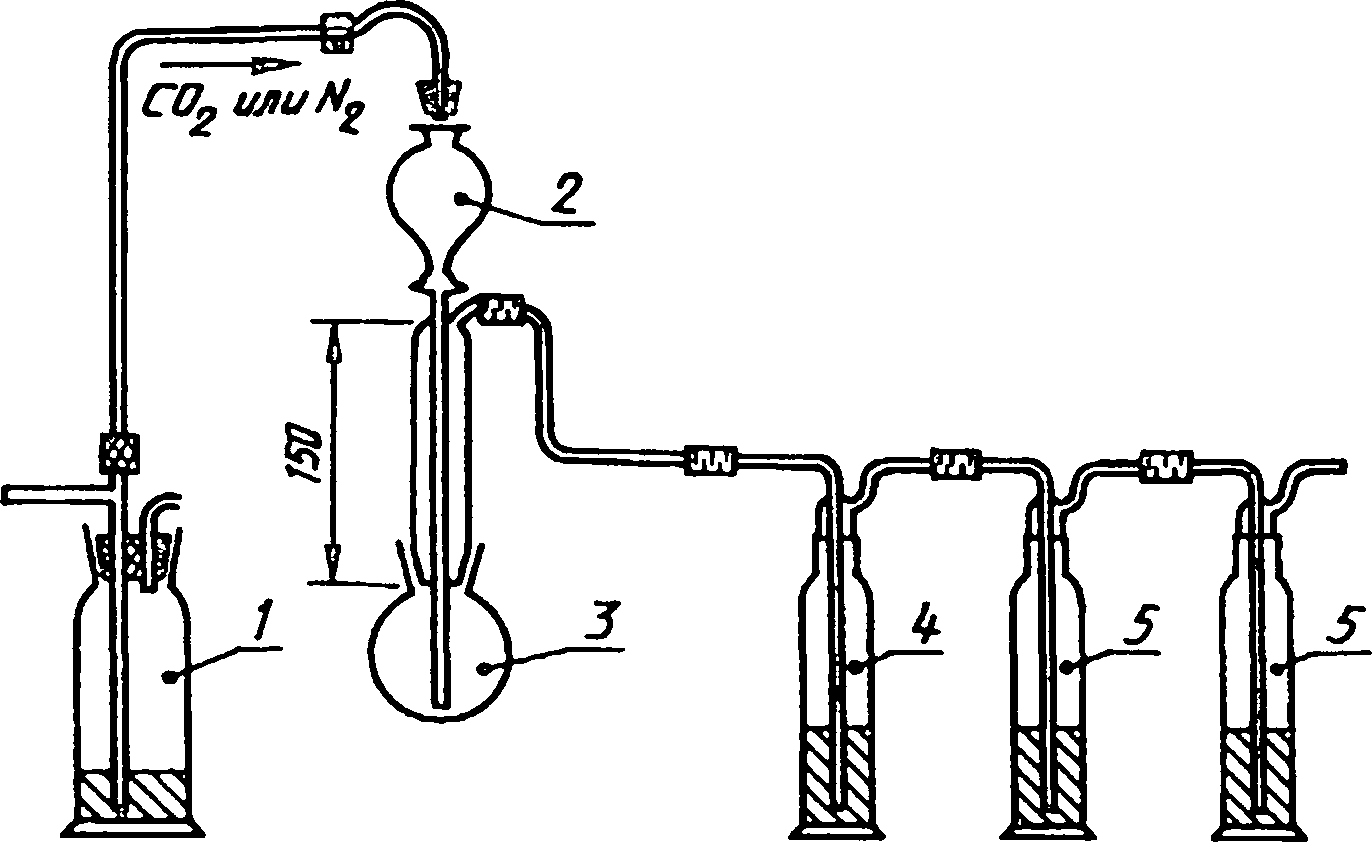
/ — ртутный затвор; 2 — капельная воронка вместимостью 80 см3;
3— реакционная колба вместимостью 100 см3; 4 — поглотительная склянка с водой; 5 — поглотительные склянки с раствором ацетата кадмия вместимостью 200 см3
Рисунок 2 — Прибор для определения пиритной серы
7.3.1 Реакционная колба круглодонная, вместимостью приблизительно 100 см3 с широким горлом и стандартным шлифом.
7.3.2 Насадка, состоящая из широкой стеклянной трубки, на нижнем конце которой имеется стандартный шлиф для соединения с колбой;
на верхнем конце трубки расположены:
а) капельная воронка, впаянная вдоль оси трубки;
б) трубка для отвода газов. Центральная часть трубки защищает от брызг и предотвращает вынос паров кислоты.
7.3.3 Ртутный затвор.
7.3.4 Поглотительная склянка типа СН-1—200 по ГОСТ 25336 с дистиллированной водой для поглощения паров соляной кислоты (50-70 см3).
7.3.5 Две поглотительные склянки типа СН-1—200 по ГОСТ 25336, наполняют (50—70 см3) раствором уксуснокислого кадмия для поглощения образующегося сероводорода.
7.3.6 Баллон со сжатым диоксидом углерода или азотом,
ТА Проведение анализа
7.4.1 5—10 г аналитической пробы дополнительно измельчают до состояния» при котором 95 % пробы имеет крупность частиц менее 0,02 мм. Измельченную пробу тщательно перемешивают.
7.4.2 Навеску тонкоизмельченного топлива (7.4.1) массой 0,500 г помещают в реакционную колбу (7.3.1) прибора, изображенного на рисунке 2.
Принимают меры для предотвращения поглощения влаги навеской при взвешивании.
7.4.3 В реакционную колбу добавляют 3—5 см3 этанола (7.2.4), 15 г цинка (7.2.3) и 0,1 г хрома (7.2.2); встряхивают колбу до тех пор, пока содержимое не станет пастообразным.
Присоединяют колбу к прибору; в капельную воронку наливают 70 см3 соляной кислоты (7.2.5), в которую добавлено 3 см3 этанола (7.2.4).
7.4.4 В реакционную колбу под давлением диоксида углерода или азота вводят 5 см3 соляной кислоты для образования Сг+2 и затем в течение 10 мин оставшуюся кислоту.
После того, как прекратится выделение водорода, в течение 15—20 мин пропускают поток диоксида углерода иди азота для удаления сероводорода из прибора в поглотительные склянки с уксуснокислым кадмием (примечание).
7.4.5 В поглотительный сосуд с осадком сульфида кадмия (примечание) добавляют определенный объем раствора йода (7.2.10) и некоторое количество в избыток до получения четко выраженной желтой окраски. Измеряют общий объем раствора йода, добавляют 20 см3 соляной кислоты (7.2.6) и интенсивно перемешивают до растворения осадка. Содержимое поглотительной склянки переносят в коническую колбу или стакан, поглотительную склянку промывают водой и добавляют промывные воды в стакан или колбу; добавляют раствор крахмала (7.2.12) и титруют избыток йода раствором тиосульфата натрия (7.2.9) до полного обесцвечивания раствора.
Примечания
1 Вторая поглотительная склянка с уксуснокислым кадмием необходима, так как при большом содержании пиритной серы в пробе для поглощения всего сероводорода недостаточно одной поглотительной склянки.
2 При помутнении раствора во втором поглотительном сосуде содержимое обоих поглотительных сосудов объединяют перед добавлением раствора йода.
3 В поглотительном сосуде с водой улавливается хлористый водород. После каждого определения воду в сосуде меняют.
4 Рекомендуемая скорость потока газа через прибор — 40 дм3/ч.
7.5 Обработка результатов
Массовую долю пиритной серы в аналитической пробе S“, %, (по массе) вычисляют по формуле
sa = (2Kt Л/6-К7 . М7) . 0.016 1(х)_ а>
где И6 — объем раствора йода, взятый для анализа, см3;
И7 — объем раствора тиосульфата натрия, израсходованного на титрование избытка йода, см3;
Мв — концентрация раствора йода, моль/дм3;
М7 — концентрация раствора тиосульфата натрия, моль/дм3;
т5 — масса навески, г;
S* — массовая доля сульфидной серы, %.
Результаты (предпочтительно среднее значение двух определений) округляют до 0,1 %.
7.6 Точность метода — по 6.7.
8 ОПРЕДЕЛЕНИЕ СУЛЬФИДНОЙ СЕРЫ
8.1 Сущность метода — по 3.4.
8.2 Реактивы
8.2.1 Требования к реактивам — по 7.2.1
8.2.2 Этанол по 7.2.4.
8.2.3 Соляная кислота по 7.2.5.
8.2.4 Соляная кислота по 7.2.6.
8.2.5 Уксусная кислота по 7.2.7.
8.2.6 Кадмий уксуснокислый по 7.2.8.
8.2.7 Натрий серноватистокислый (тиосульфат натрия), На/82О?5Н2О, раствор 0,02 моль/дм3. 4,964 г тиосульфата натрия растворяют в небольшом объеме воды в мерной колбе вместимостью 1 дм3 и доливают водой до метки.
8.2.8 Калий йодистый по ГОСТ 4232.
8.2.9 Йод по ГОСТ 4159, раствор 0,01 моль/дм3.
2,538 г йода и 5 г йодистого калия растворяют в 40 см3 воды в мерной колбе вместимостью 1 дм3 и затем доливают водой до метки. Концентрацию раствора устанавливают по раствору тиосульфата натрия (8.2.7).
8.2.10 Крахмал растворимый по 7.2.12.
8.2.11 Диоксид углерода или азот, газ по 7.2.13.
8.3 Аппаратура — по 7.3.
Примечание— Взамен ртутного затвора (7.3.3) допускается применять поглотительные склянки с водой для выравнивания давления потока газа.
8.4 Проведение анализа
8.4.1 Собирают прибор по рисунку 2. В поглотительную склянку 5 наливают 50 см3 воды, в сосуды 6 — по 50 см3 раствора уксуснокислого кадмия.
В сосуд 2 наливают воду и погружают нижний конец трубки настолько, чтобы обеспечить скорость газового потока окало 40 дм3/ч.
В капельную воронку наливают 70 см3 соляной кислоты (8.2.3).
8.4.2 Навеску топлива массой 5 г помещают в реакционную колбу (7.3.1) прибора. Добавляют 10 см3 этанола и встряхиванием перемешивают смесь до пастообразного состояния.
Колбу присоединяют к прибору и постепенно вводят соляную кислоту из капельной воронки под давлением диоксида углерода или азота.
В течение 30мин через прибор пропускают диоксид углерода или азот со скоростью 40 дм?/ч. Выделяющийся сероводород поглощается раствором уксуснокислого кадмия.
8.4.3 Подготовка к титрованию и титрование — по 7.4.5.
8.5 Обработка результатов
Массовую долю сульфидной серы в аналитической пробе топлива S*, %, вычисляют по формуле
s. = (г^лу^л/^о/иб 1(Ю
‘ % ’ ’
где V8 — объем раствора йода, взятого для анализа, см3;
V9 — объем раствора тиосульфата натрия, израсходованного на титрование избытка йода, см3;
Ms — концентрация раствора йода, моль/дм3;
М9 — концентрация раствора тиосульфата натрия, моль/дм3; т6 — масса навески, г.
Результаты (предпочтительно среднее значение двух определений) округляют до 0,1 %.
8.6 Точность метода
Таблица 3
Сульфидная сера | Максимально допустимые расхождения между результатами (рассчитанными для одинакового содержания влаги) |
в одной лаборатории (сходимость) | в разных лабораториях (воспроизводимость) |
0,05 % абс | 0,10 % абс |
8.6.1 Сходимость — по 5.6.1.
8.6.2 Воспроизводимость — по 5.6.2.
9 ВЫЧИСЛЕНИЕ МАССОВОЙ ДОЛИ ОРГАНИЧЕСКОЙ СЕРЫ
Массовую долю органической серы в аналитической пробе топлива S®, %, вычисляют по формуле
где S, — массовая доля общей серы; определяемая по ГОСТ 8606, %.
10 ПРОТОКОЛ ОПРЕДЕЛЕНИЯ
Протокол определения должен включать следующую информацию:
а) ссылку на применяемый метод;
б) результаты и способ их выражения;
в) особенности, замеченные при проведении анализа;
г) операции, не предусмотренные настоящим стандартом или необязательные.
ПРИЛОЖЕНИЕ А (справочное)
ПОЯСНЕНИЯ К ТЕРМИНАМ, ПРИМЕНЯЕМЫМ В СТАНДАРТЕ
1 Общая сера — сумма разных форм соединений серы в органической и. минеральной массе топлива.
2 Сульфатная сера — часть общей серы топлива, входящая в состав сульфатов металлов.
3 Пиритная сера — часть общей серы топлива, входящая в состав пирита, марказита и мельниковита (железных колчеданов).
4 Сульфидная сера — часть общей серы топлива, входящая в состав сульфидов металлов.
5 Органическая сера — часть общей серы топлива, входящая в состав органической массы топлива (органических соединений серы).
УДК 662.1.001.4:006.354 ОКС 73.040 А19 ОКСТУ 0309
Ключевые слова: угли бурые, угли каменные, антрацит, лигниты, горючие сланцы, кокс, сера сульфатная, сера пиритная, сера сульфидная, сера органическая, торф, испытание
Редактор Р.С. Федорова Технический редактор В.Н. Прусакова Корректор Р.А. Ментова Компьютерная верстка А.С. Юфина
Изд. лиц. № 021007 от 10.08.95. Сдано в набор 19.11.96. Подписано в печать 17.12.96. Усл.печ.л. 1,40. Уч.-изд.л. 1,30. Тираж 201 экз. С 4145. Зак. 679.
ИПК Издательство стандартов 107076, Москва, Колодезный пер., 14. Набрано в Издательстве на ПЭВМ Филиал ИПК Издательство стандартов — тип. "Московский печатник” Москва, Лялин пер., 6
1
Допускается до введения И СО 1170—77 в качестве государственного стандарта.
