
ГОСТ Р ИСО 14708-1-2012
Группа P26
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ИМПЛАНТАТЫ ХИРУРГИЧЕСКИЕ
Активные имплантируемые медицинские изделия
Часть 1
Общие требования к безопасности, маркировке и информации, предоставляемой изготовителем
Implants for surgery. Active implantable medical devices. Part 1. General requirements for safety, marking and information to be provided by the manufacturer
OКC 11.040.40
Дата введения 2014-01-01
Предисловие
1 ПОДГОТОВЛЕН Федеральным государственным бюджетным учреждением "Научный Центр сердечно-сосудистой хирургии им.А.Н.Бакулева" РАМН (ФГБУ "НЦССХ им.А.Н.Бакулева" РАМН) на основе собственного аутентичного перевода на русский язык международного стандарта, указанного в пункте 4
2 ВНЕСЕН Управлением технического регулирования и стандартизации Федерального агентства по техническому регулированию и метрологии
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 30 августа 2012 г. N 265-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 14708-1:2000* "Имплантаты хирургические. Активные имплантируемые медицинские изделия. Часть 1. Общие требования к безопасности, маркировке и информации, предоставляемой изготовителем" (ISO 14708-1:2000 "Implants for surgery - Active implantable medical devices - Part 1: General requirements for safety, marking and information to be provided by the manufacturer").
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012 (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте национального органа Российской Федерации по стандартизации в сети Интернет (gost.ru)
1 Область применения
Настоящий стандарт устанавливает общие требования к активным имплантируемым медицинским изделиям.
Примечание - Для определенных типов активных имплантируемых медицинских изделий настоящие требования дополняются требованиями специальных стандартов, которые формируют составные части ИСО 14708. Особое внимание следует уделить применению настоящего стандарта в отношении тех активных имплантируемых медицинских изделий, для которых не существует специальных стандартов.
Методы испытаний, приведенные в настоящем стандарте, предназначены для оценки соответствия на образцах изделий и не предназначены для испытаний изготавливаемой продукции.
Настоящий стандарт применим к активным имплантируемым медицинским изделиям, приводимых в действие не только силой электрического тока, но и с помощью других источников энергии (например, давления газа или пружинного механизма).
Настоящий стандарт также применим к некоторым видам неимплантируемых частей медицинских изделий и принадлежностей к ним (см. 3.3).
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на нижеследующие стандарты*. Для датированных ссылок применяют только указанные издания. Для недатированных ссылок применяют самые последние издания (включая любые изменения и поправки).
_______________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ИСО 8601:1998 Элементы данных и форматы для обмена информацией. Обмен информацией. Представление дат и времени (ISO 8601:1998, Data elements and interchange formats - Information interchange - Representation of dates and times)
ИСО 11607:1997 Упаковка для медицинских изделий, подлежащих финишной стерилизации (ISO 11607:1997, Packaging for terminally sterilized medical devices)
ИСО 14155:1996 Клинические исследования медицинских изделий (ISO 14155:1996, Clinical investigation of medical devices)
ИСО 15223:2000 Изделия медицинские. Символы, используемые на этикетках и ярлыках медицинских изделий и в предоставляемой информации (ISO 15223:2000, Medical devices - Symbols to be used with medical devices labels, labeling and information to be supplied)
МЭК 60068-2-14:1986 Испытание на воздействие внешних факторов. Часть 2. Испытания. Испытание N: Изменение температуры (IEC 60068-2-14:1986, Environmental testing - Part 2: Tests N: Change of temperature)
МЭК 60068-2-32:1990 Испытания на воздействия внешних факторов. Часть 2. Испытания. Испытание Ed: Свободное падение (Процедура 1) [IEC 60068-2-32:1990, Environmental testing - Part 2: Tests Ed: Free fall (Procedure 1)]
МЭК 60068-2-47:1999 Испытания на воздействие внешних факторов. Часть 2-47. Методы испытаний. Крепление элементов, аппаратуры и других изделий для испытаний на вибрацию, удар и для подобных динамических испытаний (IEC 60068-2-47:1999, Environmental testing - Part 2-47: Test methods - Mounting of components, equipment and other articles for vibration, impact and similar dynamic tests)
МЭК 60068-2-64:1993 Испытания на воздействие внешних факторов. Часть 2. Испытания. Испытание Fh: Широкополосная случайная вибрация (цифровое управление) и руководство [IEC 60068-2-64:1993, Environmental testing - Part 2: Tests methods - Test Fh: Vibration, broad-band random (digital control) and guidance)]
МЭК 60601-1:1988 Медицинское электрооборудование. Часть 1. Общие требования безопасности. Дополнение 1:1991 и Дополнение 2:1995 (IEC 60601-1-1:1988, Medical electrical equipment - Part 1: General requirements for safety. Amendment 1:1991 and Amendment 2:1995)
МЭК 60601-1-1:1992 Медицинское электрооборудование. Часть 1. Общие требования безопасности. 1. Дополняющий стандарт. Требования безопасности к медицинским электрическим системам (IEC 60601-1-1:1992, Medical electrical equipment - Part 1: General requirements for safety - 1. Collateral standard: Safety requirements for medical electrical systems)
МЭК 60601-1-2:1993 Медицинское электрооборудование. Часть 1. Общие требования безопасности. 2. Дополняющий стандарт. Электромагнитная совместимость. Требования и испытания (IEC 60601-1-2:1993, Medical electrical equipment - Part 1: General requirements for safety - 2. Collateral standard: Electromagnetic compatibility - Requirements and tests)
МЭК 60601-1-4:1996 Медицинское электрооборудование. Часть 1. Общие требования безопасности. 4. Дополняющий стандарт. Программируемые медицинские электрические системы (IEC 60601-1-4:1996, Medical electrical equipment - Part 1: General requirements for safety - 4. Collateral standard: Programmable electrical medical systems)
МЭК 60601-2-27:1994 Медицинское электрооборудование. Часть 2. Специальные требования безопасности электрокардиографического мониторингового оборудования (IEC 60601-2-27:1994, Medical electrical equipment - Part 2: Particular requirements for the safety of electrocardiographic monitoring equipment)
МЭК 61000-4-2:1995 Электромагнитная совместимость. Часть 4. Методы испытаний и измерений. Секция 2. Испытание на воздействие электростатического разряда. Основные публикации по электромагнитной совместимости (IEC 61000-4-2:1995, Electromagnetic compatibility (EMC) - Part 4: Testing and measurement techniques - Section 2: Electrostatic discharge immunity test. Basic EMC Publication)
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 медицинское изделие: Изделие вместе с принадлежностями или программным обеспечением, необходимым для его корректного функционирования, предназначенное изготовителем для клинического применения самостоятельно или в сочетании с другими изделиями с целью:
- диагностики, профилактики, мониторинга, лечения или облегчения течения заболеваний или последствий травм;
- исследования, замещения или изменения анатомии или физиологического процесса;
- контроля зачатия,
при условии, что его основное предназначение достигается не за счет фармакологических, иммунологических или метаболических средств, но его функционирование может поддерживаться такими средствами.
3.2 активное медицинское изделие: Медицинское изделие, функционирующее за счет электрической энергии или любого другого источника энергии, за исключением сил гравитации или энергии, непосредственно генерируемой телом человека.
3.3 активное имплантируемое медицинское изделие: Активное медицинское изделие, предназначенное для полного или частичного введения в организм человека хирургическим или медикаментозным путем, либо путем введения через анатомические отверстия тела и для которого предусмотрено, что оно остается на месте после данной процедуры.
Примечание - В рамках настоящего стандарта активным имплантируемым медицинским изделием может быть как однокомпонентное активное медицинское изделие, так и многокомпонентная система, содержащая набор компонентов и принадлежностей, которые, взаимодействуя, достигают уровня функционирования, предусмотренного изготовителем. Не все эти компоненты или принадлежности могут требовать полной или частичной имплантации, но необходимо определить специальные требования к неимплантируемым компонентам и принадлежностям, если таковые могут повлиять на безопасность или эффективность имплантируемого устройства.
3.4 катетер: Гибкая трубка, позволяющая получить доступ к определенной зоне внутри тела человека через сосудистый просвет (или какого-либо препарата).
Примечание - Катетер может быть соединен с проводником.
3.5 проводник: Гибкая трубка, содержащая один или несколько изолированных от внешней среды электропроводников, предназначенная для передачи электроэнергии по ее длине.
Примечание - Проводник может быть соединен с катетером.
3.6 одноразовая упаковка: Упаковка одноразового использования, сконструированная таким образом, чтобы ее содержимое могло быть стерилизовано и сохраняло стерильность.
3.7 стерильная упаковка: Одноразовая упаковка, в которой было простерилизовано содержимое.
3.8 товарная упаковка: Упаковка, защищающая и идентифицирующая активное имплантируемое медицинское изделие во время хранения и обращения с ним.
Примечание - Товарная упаковка может быть вложена в другую упаковку, например, в "транспортную тару" для транспортирования.
3.9 маркировка: Надпись на изделии, упаковке или этикетке.
3.10 этикетка: Наклейка, используемая для нанесения маркировки, крепящаяся к изделию или упаковке, но не являющаяся неотъемлемой частью изделия или упаковки.
3.11 радиоактивное вещество: Любое вещество, содержащее один и более нуклидов, активностью или концентрацией которых нельзя пренебрегать, если дело касается радиационной защиты.
3.12 закрытый источник излучения: Источник, содержащий радиоактивные вещества, которые помещены в надежный твердый и эффективно инактивированный материал или опечатаны в инактивированном контейнере, прочность которого достаточна для предотвращения любых утечек радиоактивных веществ при нормальных условиях эксплуатации.
3.13 лекарственное средство: Средство, которое при самостоятельном использовании предназначено для лечения или предотвращения заболеваний или которое может быть введено с целью диагностики, восстановления, корректировки или изменения физиологических функций человека.
3.14 вред: Физическая травма или ущерб здоровью людей или имуществу.
3.15 опасность: Потенциальный источник вреда.
3.16 неприемлемая опасность: Опасность, вероятность причинения вреда при которой выше, чем установленное значение, определяемое тяжестью причинения вреда.
3.17 контролируемая опасность: Основная конструктивная характеристика активного имплантируемого медицинского изделия, призванная обеспечить надежность того, что изделие не станет причиной возникновения неприемлемой опасности.
3.18 портативное оборудование: Оборудование, для которого предусмотрено перемещение из одного положения в другое во время использования или между периодами использования при переноске одним или более лицом.
3.19 ручное оборудование: Оборудование, которое при нормальном использовании предусматривает ручной способ обращения и удерживания.
4 Символы и аббревиатуры
На маркировке активного имплантируемого медицинского изделия или сопроводительной документации к нему, по возможности, следует применять символы, аббревиатуры и цветовую идентификацию. Символы, аббревиатуры и цветовая идентификация должны соответствовать действующим международным стандартам и соглашениям (например, ИСО 15223). В случае если не существует стандартов, то символы, аббревиатуры и цветовая идентификация должны быть описаны в сопроводительной документации.
Соответствие маркировки проверяют в ходе осмотра.
Примечание - Символы, используемые для активных имплантируемых медицинских изделий определенных видов, могут быть уточнены в последующих частях ИСО 14708.
5 Общие требования к неимплантируемым частям
Неимплантируемые части активного имплантируемого медицинского изделия, подсоединяемые к источнику энергии или оснащенные таковым, должны соответствовать требованиям МЭК 60601-1, МЭК 60601-1-1, МЭК 60601-1-2 и МЭК 60601-1-4, за исключением случаев, когда какое-либо из требований в этих стандартах заменено требованием настоящего стандарта.
Примечание 1 - МЭК 60601-1 применяется здесь в том виде, в каком он применялся бы для электромедицинского изделия, для которого нет специализированного стандарта. В настоящем стандарте ссылаются на отдельные разделы МЭК 60601-1, если они касаются фундаментальных положений ИСО 14283.
Примечание 2 - Требования к неимплантируемым частям активных имплантируемых медицинских изделий определенных видов могут быть уточнены в последующих частях ИСО 14708.
6 Требования к активным имплантируемым медицинским изделиям определенных видов
Требования к активным имплантируемым медицинским изделиям определенных видов не излагаются подробно в настоящем стандарте, но могут быть уточнены в последующих частях ИСО 14708.
7 Общие требования к упаковке
7.1 Имплантируемые части активного имплантируемого медицинского изделия следует поставлять в одноразовой упаковке (см. 14.1).
Одноразовая упаковка должна быть разработана таким образом, чтобы позволять изготовителю стерилизовать уже запечатанное содержимое.
Соответствие проверяют в ходе осмотра.
7.2 Одноразовая упаковка должна быть вложена в товарную упаковку.
Соответствие проверяют в ходе осмотра.
8 Общая маркировка активных имплантируемых медицинских изделий
Примечание 1 - См. также раздел 4.
8.1 Любые предупреждающие надписи, требуемые настоящим стандартом, должны быть четко выполнены и различимы.
Соответствие проверяют в ходе осмотра.
8.2 Имплантируемые части изделий и компоненты этих частей должны быть идентифицированы таким образом, чтобы не препятствовать выполнению всех необходимых измерений, проводимых для выявления возможной опасности, связанной с любой имплантируемой частью.
Соответствие подтверждают анализом документации изготовителя, поясняющей связь между идентификацией активного имплантируемого медицинского изделия и его частей.
9 Маркировка товарной упаковки
9.1 Если товарная упаковка содержит радиоактивные вещества, на нее должна быть нанесена маркировка, указывающая вид и активность радиоактивного вещества.
Соответствие проверяют в ходе осмотра.
9.2 На товарной упаковке должны быть указаны адрес и наименование изготовителя, включая, по крайней мере, город и страну.
Соответствие проверяют в ходе осмотра.
9.3 На товарную упаковку должно быть нанесено наименование изделия (например, электрокардиостимулятор), модель, назначение изделия, номер партии или серийный номер изделия.
Соответствие проверяют в ходе осмотра.
9.4 Товарная упаковка имплантируемых частей активного имплантируемого медицинского изделия должна содержать любую дополнительную информацию и, если необходимо, соответствующие характеристики для идентификации изделия.
Соответствие проверяют в ходе осмотра.
9.5 Товарная упаковка имплантируемых частей активного имплантируемого медицинского изделия должна содержать подтверждение того, что содержимое упаковки было стерилизовано.
Соответствие проверяют в ходе осмотра.
9.6 На товарной упаковке имплантируемых частей активного имплантируемого медицинского изделия должны быть указаны год и месяц изготовления, в цифровом выражении, согласно ИСО 8601.
Соответствие проверяют в ходе осмотра.
9.7 На товарной упаковке имплантируемых частей активного имплантируемого медицинского изделия должна быть указана дата "использовать до" (год и месяц).
Соответствие проверяют в ходе осмотра.
9.8 Маркировка на товарной упаковке имплантируемых частей активного имплантируемого медицинского изделия должна идентифицировать содержимое внутри упаковки или, если места на товарной упаковке недостаточно, сведения о содержимом должны быть приведены внутри упаковки.
Соответствие проверяют в ходе осмотра.
9.9 Если согласно назначению имплантируемых частей активного имплантируемого медицинского изделия, вложенных в товарную упаковку, требуется, чтобы они подключались к другому изделию или принадлежностям, не включенным в товарную упаковку, на товарной упаковке должны быть обозначены тип или конфигурация требуемого соединителя.
Соответствие проверяют в ходе осмотра.
9.10 На товарной упаковке имплантируемых частей активного имплантируемого медицинского изделия должно быть четкое указание предполагаемого применения изделия, если оно не очевидно из описания изделия, требуемого 9.3 и 9.4.
Соответствие проверяют в ходе осмотра.
9.11 На товарную упаковку должна быть нанесена информация о любых специальных условиях окружающей среды или ограничениях по транспортированию (например, беречь от ударов, вибрации, температуры, давления или влажности), необходимая для корректного транспортирования и хранения изделия.
Соответствие проверяют в ходе осмотра.
9.12 Если изделие предназначено для определенного использования, товарная упаковка должна содержать указание на специальные виды использования (например, "изготовлено на заказ" или "только для клинических исследований").
Соответствие проверяют в ходе осмотра.
10 Конструкция товарной упаковки
10.1 Товарная упаковка активного имплантируемого медицинского изделия должна быть изготовлена так, чтобы защитить изделие и противостоять нанесению вреда от падения (удара), укладывания в стопку (сдавливания), вибрации и температуры, которые могут возникнуть при хранении или транспортировании, согласно указаниям изготовителя.
Соответствие проверяют путем осмотра и анализа отчетных данных, предоставленных изготовителем.
10.2 Товарная упаковка активного имплантируемого медицинского изделия должна быть достаточно защищена от воздействия влаги во время хранения или транспортирования для предотвращения видимой порчи упаковки, маркировок, этикеток или эксплуатационной документации.
Методика тестирования. Помещают товарную упаковку в испытательную камеру на двое суток. Температура в испытательной камере должна быть стабильна и составлять (30±2) °С. Относительная влажность в испытательной камере должна быть (93±3)%.
Соответствие проверяют анализом записей изготовителя.
10.3 Маркировка на товарной упаковке активного имплантируемого медицинского изделия должна быть несмываемой.
Методика тестирования. Испытуемую упаковку кладут таким образом, чтобы маркировка при испытании располагалась горизонтально в самом верхнем положении. Разбрызгивают 10 мл воды над центральной частью упаковки. Через одну минуту следует промокнуть маркировку, используя сухую мягкую ткань.
Соответствие считается подтвержденным, если после проведения процедуры, описанной выше, вся маркировка остается четко различимой. При нанесении маркировки на клейкую этикетку клейкая фиксация этикетки не должна ослабнуть и этикетка не должна изогнуться ни с одной из сторон.
10.4 Товарная упаковка должна обеспечивать соответствие между содержимым - собственно активным имплантируемым медицинским изделием - и информацией в эксплуатационной документации, определяющей назначение и функции изделия, а также условия выполнения имплантации.
Соответствие проверяют в ходе осмотра.
11 Маркировка стерильной упаковки
11.1 Маркировка стерильной упаковки должна содержать наименование изготовителя и адрес (город и страна) производства.
Соответствие проверяют в ходе осмотра.
11.2 На стерильной упаковке должно быть указание, что упаковка и ее содержимое были стерилизованы; также должен быть указан метод стерилизации (см. ИСО 15223 в части рекомендуемых символов).
Соответствие проверяют в ходе осмотра.
11.3 На стерильной упаковке должен быть отчетливо различим символ
![]()
в соответствии с ИСО 15223.
Соответствие проверяют в ходе осмотра.
11.4 На стерильной упаковке должны быть указаны год и месяц, когда упакованное изделие было изготовлено, согласно требованиям 9.6.
Соответствие проверяют в ходе осмотра.
11.5 На стерильной упаковке должна быть указана дата "использовать до" согласно требованиям 9.7. Соответствие проверяют в ходе осмотра.
11.6 Стерильная упаковка должна содержать описание изделия согласно требованиям 9.3. Соответствие проверяют в ходе осмотра.
11.7 Маркировка стерильной упаковки должна идентифицировать содержимое, за исключением случаев, когда стерильная упаковка прозрачна и ее содержимое можно увидеть.
Соответствие проверяют в ходе осмотра.
11.8 Если согласно предполагаемому применению изделия, вложенного в стерильную упаковку, требуется, чтобы оно было подключено к другому изделию или принадлежностям, не входящим в содержимое стерильной упаковки, на последней должен быть обозначен тип или приведена схема требуемого типа соединителя согласно требованиям 9.9.
Соответствие проверяют в ходе осмотра.
11.9 Стерильная упаковка должна содержать инструкцию по ее открытию.
Соответствие проверяют в ходе осмотра.
11.10 Если изделие предусмотрено для специального использования, на стерильной упаковке должно быть указание на специальные цели (например, "изготовлено на заказ" или "только для клинических исследований").
Соответствие проверяют в ходе осмотра.
12 Конструкция одноразовой упаковки
12.1 Одноразовая упаковка должна соответствовать требованиям ИСО 11607.
Соответствие проверяют контролем и анализом записей, предоставленных изготовителем.
12.2 Одноразовая упаковка должна быть разработана таким образом, чтобы, если упаковка однажды была вскрыта, это было бы явно видно, и даже если она была открыта и потом запечатана снова, все равно было бы заметно, что упаковка открывалась.
Соответствие проверяют в ходе осмотра.
12.3 Маркировка на одноразовой упаковке должна быть несмываемой.
Соответствие проверяют, как указано в 10.3.
13 Маркировка активных имплантируемых медицинских изделий
13.1 Насколько это практично и необходимо, на активном имплантируемом медицинском изделии должны быть указаны наименование и торговая марка изготовителя, обозначение модели изделия и, если применимо, номер партии или серийный номер изделия.
Соответствие проверяют в ходе осмотра и путем испытания нанесенных обозначений на влажное истирание с применением влагосодержащего средства.
Методика тестирования на истирание с помощью влагосодержащего средства. Маркировку изделия на упаковке следует потереть вручную, без излишнего давления, сначала лоскутом ткани в течение 15 с, смоченным в метиловом спирте температурой, равной температуре окружающей среды, а затем в течение 15
с лоскутом ткани, смоченным в воде температурой, равной температуре окружающей среды, после чего маркировка должна сохранить четкость и различимость.
13.2 Если отдельные имплантируемые части конкретной модели активного имплантируемого медицинского изделия содержат различные источники энергии, то должна быть предусмотрена возможность группировать изделия по виду источника энергии, например, путем ссылки на инструкцию по применению или используя соответствующий индекс в обозначениях.
Соответствие проверяют в ходе осмотра.
13.3 Имплантируемые части активного имплантируемого медицинского изделия с внутренним источником энергии должны иметь код, по которому изделие и изготовитель могут быть однозначно идентифицированы (особенно в отношении модели изделия). В случае отсутствия специальных знаний о марке или модели изделия должна быть предусмотрена возможность считывания этого кода.
Соответствие проверяют способом, указанным изготовителем в инструкции по применению (см. 28.6).
13.4 Любые визуальные индикаторы, имеющиеся на активном имплантируемом медицинском изделии, должны быть понятными для пользователя с помощью эксплуатационной документации, учитывая уровень его знаний.
Соответствие проверяют в ходе осмотра.
14 Защита от непреднамеренных биологических воздействий, вызванных активным имплантируемым медицинским изделием
14.1 Любая имплантируемая часть активного имплантируемого медицинского изделия или другие его части, вложенные в одноразовую упаковку (см. 7.1) и не находящиеся внутри имплантируемого герметично изолированного контейнера, должны быть стерильны.
Соответствие считается подтвержденным, если в процессе валидации записей, предоставленных изготовителем, устанавливается, что одноразовая упаковка была стерилизована с использованием валидированного процесса (например, в соответствии с ИСО 11134 или ISO 11135).
14.2 Для любой части активного имплантируемого медицинского изделия, для которой при нормальном использовании предполагается контакт с жидкими средами организма человека, не допускается выделений твердых частиц в неприемлемом количестве при условии соблюдения правил использования изделия согласно указаниям изготовителя.
Методика тестирования. Асептически извлекают активное имплантируемое медицинское изделие из одноразовой упаковки. Погружают имплантируемую часть в раствор соли 9 г/л, пригодный для инъекций, в нейтральном стеклянном контейнере. Объем соляного раствора должен быть примерно в 10-20 раз больше объема испытуемой части. Накрывают контейнер стеклянной пластиной и оставляют при температуре примерно (37±2) °С на 8-18 ч, периодически встряхивая ванночку. Готовят контрольный раствор схожего объема с той же солью, поддерживают те же условия, что и в ванночке с образцом. Сравнивают образцы жидкости из ванночки с образцом из контрольной ванночки, применяя подходящую аппаратуру для автоматического измерения размеров частиц, работающую по принципу электрического зонного зондирования (например, принцип Коултера), применяя процедуру отбора образцов, рекомендованную изготовителем.
Соответствие считается подтвержденным, если среднее число частиц, выделившихся из образца, по отношению к таковому для контрольного образца не превышает 1000 частиц/мл для частиц размерами более 2,0 мкм и не превышает 100 частиц/мл для частиц размерами более 5,0 мкм.
14.3 Части изделия, предназначенные для прокалывания кожи человека, должны быть биосовместимы.
Соответствие считается подтвержденным, если записи, предоставленные изготовителем, подтверждают, что биосовместимость выбранных материалов была продемонстрирована:
a) по аналогии с опубликованными данными, или
b) за счет того, что выбранный материал уже подтвердил биосовместимость в ранее проведенных клинических испытаниях в подобном применении, или
c) за счет опыта использования подобного изделия, уже существующего на рынке, вместе с доказательствами контроля материалов, применяемых при изготовлении изделий, или
d) за счет соответствия опубликованным процедурам биологической оценки медицинских изделий.
14.4 Если активное имплантируемое медицинское изделие содержит лекарственное вещество и это вещество или его производная предназначены для введения в тело пациента (хотя само вещество является составной частью активного имплантируемого медицинского изделия), такое вещество должно быть как безопасным, так и эффективным относительно заявленной функции активного имплантируемого медицинского изделия.
Соответствие считается подтвержденным, если записи, предоставленные изготовителем, подтверждают, что безопасность и качество лекарственного препарата были проверены с использованием надлежащих методов.
15 Защита пациента или пользователя от вреда, вызванного внешними физическими особенностями активного имплантируемого медицинского изделия
15.1 Наружные поверхности неимплантируемых частей активного имплантируемого медицинского изделия должны соответствовать требованиям МЭК 60601-1, раздел 23.
Соответствие проверяют в ходе осмотра.
15.2 Поверхности имплантируемых частей активного имплантируемого медицинского изделия не должны иметь никаких особенностей: ни острых углов или краев, которые могут вызвать избыточную реакцию или раздражение, превышающее пределы реакций, вызванных имплантацией, ни грубо обработанных поверхностей, которые не требуются для правильного функционирования изделия.
Соответствие проверяют в ходе осмотра.
16 Защита пациента от вреда, вызванного электрическим током
16.1 Внешние части активного имплантируемого медицинского изделия должны соответствовать требованиям МЭК 60601-1, раздел 19.
Соответствие подтверждают согласно МЭК 60601-1.
16.2 За исключением предусмотренных функций, имплантируемые части активного имплантируемого медицинского изделия, содержащие источник энергии, должны быть электрически нейтральны при контакте с телом человека. При использовании изделия ни в одной из линий тока не должно появляться утечек тока (прямого тока) более 1 мкА.
Соответствие подтверждают контролем процедур данного испытания и анализом результатов, предоставленных изготовителем.
16.3 Изолированные части имплантируемых питающих проводов или катетеров со встроенными электрическими проводниками, которые подвергаются воздействию разницы электрических потенциалов более 10 Вт, должны выдерживать испытание на диэлектрическое сопротивление, при котором прикладываемая разность потенциалов должна быть не менее удвоенной пиковой разности потенциалов, испытываемой этой изолированной частью.
Методика тестирования. Проводят предварительную подготовку изолированных частей имплантируемых питающих проводов или катетеров со встроенными электрическими проводниками, используя полное погружение в раствор соли 9 г/л при температуре (37±2) °C минимум на 10 суток. После промывки испытуемой части дистиллированной водой и протирки поверхности от воды испытывают эту часть на диэлектрическое сопротивление методом, указанным изготовителем.
Соответствие проверяют контролем процедур данного испытания и анализом результатов, предоставленных изготовителем.
17 Защита пациента от вреда, вызванного тепловыделениями
Ни одна из наружных поверхностей имплантируемой части активного имплантируемого медицинского изделия не должна нагреваться более чем на 2 °С выше нормальной температуры тела пациента как при нормальной работе активного имплантируемого медицинского изделия, так и в условиях единичного отказа (см. 19.3).
Соответствие подтверждают анализом дизайна конструкции, представленного изготовителем, подтвержденного его расчетами и результатами испытаний в соответствующей форме.
Примечание - Требования по защите от тепловыделений, вызванных нагревом активных имплантируемых медицинских изделий определенных видов, могут быть уточнены в последующих частях ИСО 14708.
18 Защита от ионизирующего излучения, выделяемого или испускаемого активным имплантируемым медицинским изделием
18.1 Если активное имплантируемое медицинское изделие содержит какое-либо радиоактивное вещество, то последнее должно находиться в виде опечатанного источника излучения.
Соответствие подтверждают контролем анализа дизайна конструкции, представленного изготовителем и подтвержденного результатами испытаний в соответствующей форме.
18.2 Если активное имплантируемое медицинское изделие содержит какое-либо радиоактивное вещество, то последствия воздействия ионизирующего излучения должны быть оправданы преимуществами использования радиоактивной субстанции.
Соответствие подтверждают расчетами изготовителя и результатами испытаний в соответствующей форме.
18.3 Если активное имплантируемое медицинское изделие содержит какое-либо радиоактивное вещество, то последствия от воздействия ионизирующего излучения должны быть ограничены на возможном низком уровне.
Соответствие подтверждают контролем анализа дизайна конструкции, представленного изготовителем и подтвержденного результатами испытаний в соответствующей форме.
19 Защита от непреднамеренных воздействий, вызванных изделием
Примечание - См. также 28.20.
19.1 Имплантируемые части активного имплантируемого медицинского изделия должны быть разработаны таким образом, чтобы постепенно накапливаемые изменения в материале, которые могут возникнуть во время срока службы изделия, не вызвали неприемлемого вреда.
Методика оценки. Возможность проявления постепенных долгосрочных изменений в материале следует оценить путем применения следующих способов:
a) проведения аналогии с опубликованными данными, или
b) установленными данными о том, что выбранный материал уже подтвердил свою стабильность в проведенных клинических испытаниях при подобном применении, или
c) данными опыта применения подобного изделия, уже существующего на рынке, вместе с доказательствами контроля материалов, применяемых в изделиях, или
d) оценкой соответствия/несоответствия свойств материала согласно опубликованным процедурам оценки имплантируемых материалов.
Документированный анализ опыта применения данного материала и его роли в изделии должен идентифицировать любые опасности и показывать, что неприемлемые риски исключены.
Соответствие подтверждает изготовитель путем обзора соответствующей документации.
19.2 Если имплантируемые части активного имплантируемого медицинского изделия содержат в себе источник энергии, например, батареи, активное имплантируемое медицинское изделие в своем составе должно иметь индикатор рекомендуемой замены, который заранее предупреждает о разряжении источника энергии, что вызывает окончание срока службы изделия. Изготовитель должен определить интервал между активацией этого индикатора рекомендуемой замены и окончанием срока службы изделия.
Соответствие подтверждают в ходе анализа дизайна конструкции, представленного изготовителем и подтвержденного результатами испытаний в соответствующей форме.
19.3 Активное имплантируемое медицинское изделие должно быть разработано таким образом, чтобы отказ любого из компонентов, части изделия или (если изделие имеет встроенную электронную систему программирования) программного обеспечения не приводил к неприемлемой опасности.
Методика оценки. Идентифицируют опасности, вызываемые условиями возможного единичного отказа, ассоциирующиеся с каждой функцией изделия. Для каждой опасности оценивают вероятность вреда путем анализа конструкции с учетом любого метода контроля опасности и допущением того, что вероятность вреда, вызванного каждым из отказов, должна быть оценена. Анализ конструкции должен сопровождаться проведением испытаний соответствующего вида.
Каждый вид опасности и оценка вероятности вреда, причиняемого ею, должны быть документированы наряду с анализом конструкции и результатами соответствующих испытаний.
Соответствие подтверждают анализом обзора соответствующей документации, подготовленным изготовителем.
19.4 Возможные побочные эффекты, возникающие при предполагаемом применении активного имплантируемого медицинского изделия, не должны вызывать чрезмерного вреда.
Методика оценки. Определяют побочные эффекты и преимущества при предполагаемом применении изделия или ссылаются на результаты текущей клинической практики при сходстве изделий либо на клинические исследования, проведенные согласно требованиям ИСО 14155.
Соответствие подтверждают оценкой документации изготовителя.
19.5 Если имплантируемые части активного имплантируемого медицинского изделия предназначены для введения лекарственных средств, то изделие должно быть разработано и изготовлено таким образом, чтобы быть совместимым с предполагаемым лекарственным средством.
Соответствие подтверждают анализом дизайна изделия, представленного изготовителем, подтвержденного расчетами и данными испытаний в соответствующей форме.
20 Защита изделия от повреждений, вызванных применением наружных дефибрилляторов
Примечание - См. также 28.12.
20.1 Схема электропитания функционирующих неимплантируемых частей активного имплантируемого медицинского изделия, подсоединенных к электродам электрокардиографа, должна быть разработана таким образом, чтобы при проведении дефибрилляции последняя не влияла на изделие, при условии, что электроды дефибриллятора не вступают в прямой контакт с электродами электрокардиографа.
Соответствие проверяют в ходе выполнения процедуры и путем испытания согласно МЭК 60601-2-27, подраздел 51.101.
20.2 Части активного имплантируемого медицинского изделия, предусмотренные для имплантации в тело пациента, должны быть разработаны таким образом, чтобы дефибрилляция пациента не влияла постоянно на изделие, обеспечивая это тем, что электроды дефибриллятора не вступают в прямой контакт с имплантированными частями.
Методика тестирования. Используют генератор импульсов дефибрилляции, содержащий RCL контур, как показано на рисунке 1, с параметрами:
330 мкФ ±16,5 мкФ;
13,3 мГн ±0,13 мГн;
![]() 10 Ом ±0,2 Ом,
10 Ом ±0,2 Ом,
где - сопротивление на индуктивности, Ом;
- сопротивление импульсов дефибрилляции, Ом;
- напряжение на выходе, Вт.
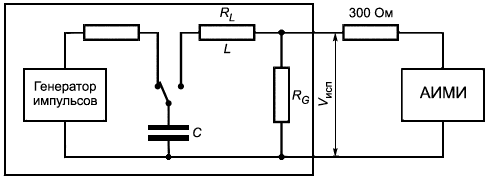
Рисунок 1 - RCL контур для оценки дефибрилляции
Максимальная амплитуда импульса напряжения на выходе генератора импульсов дефибрилляции через
должна быть (140±7) Вт.
Следует убедиться, что катушка индуктивности при импульсе магнитно не насыщена.
Определяют каждую токопроводящую часть, отличную от металлического корпуса, которая может контактировать с тканями человека. Подсоединяют генератор импульсов дефибрилляции через сопротивление (300±6) Ом (см. рисунок 1) по очереди между каждой токопроводящей частью и металлическим корпусом. Если корпус изделия заключен в металлический кожух, покрытый изолирующим материалом или изготовленный целиком из изолирующего материала, погружают корпус изделия в металлический сосуд, заполненный физиологическим раствором соли 9 г/л, и соединяют кожух с сосудом.
Испытывают каждую проводящую часть, подавая последовательность трехвольтовых импульсов положительной полярности с интервалами, равными 20 с. Затем с интервалом 60
с повторяют импульсы при отрицательной полярности (см. рисунок 2).
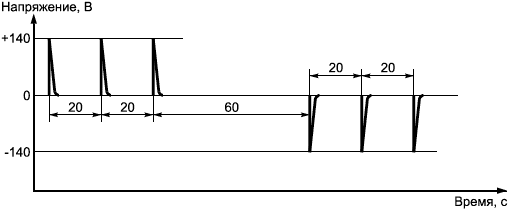
Рисунок 2 - Форма импульсов, используемых при дефибрилляции
Соответствие считается подтвержденным, если активное имплантируемое медицинское изделие соответствует спецификации изготовителя после испытания, приведенного выше.
21 Защита изделия от изменений, вызванных электрическими полями высокой энергии, приложенными непосредственно к пациенту
Имплантируемые электропроводящие части активного имплантируемого медицинского изделия, контактирующие с телом человека, должны быть сконструированы таким образом, чтобы влияние, вызванное воздействием высокоэнергетического электрического поля, приложенного непосредственно к пациенту, например, при проведении диатермии, не привело к повреждению изделия при условии, что имплантируемые части не располагаются непосредственно в зоне, по которой проходит ток, и не находятся в той части тела человека, которая является зоной лечебного воздействия.
Соответствие подтверждают анализом дизайна конструкции, представленного изготовителем, подтвержденного расчетами на основании результатов исследования в соответствующей форме.
Примечание 1 - См. также 28.12 и 28.13.
Примечание 2 - Требования по защите от изменений, вызванных высокоэнергетическими электрическими полями для активных имплантируемых медицинских изделий особых видов, могут быть уточнены в последующих частях ИСО 14708.
22 Защита активного имплантируемого медицинского изделия от изменений, вызванных применением разнородных видов лечения
Имплантируемые части активного имплантируемого медицинского изделия должны быть разработаны таким образом, чтобы воздействие ультразвуковой энергии на диагностическом уровне не могло вызвать какие-либо необратимые изменения.
Методика тестирования. Погружают имплантируемые части активного имплантируемого медицинского изделия (отличные от проводников или катетеров) в водяную ванну при комнатной температуре и в течение 1 ч подвергают воздействию ультразвуковой энергии мощностью (500±25) Вт/м, используя при этом пространственные пики и средний временной режим. Используемый сигнал должен пульсировать с коэффициентом заполнения (50±10)%. Частота должна быть выбрана от 2 до 5 МГц.
Примечание 1 - Настоящее испытание не относится к проводникам и катетерам, поскольку подразумевается, что эти изделия не будут подвергаться воздействию ультразвука в диагностическом диапазоне.
Соответствие подтверждают проверкой отсутствия необратимых изменений, вызванных испытанием, проверкой документации, предоставленной изготовителем, подтвержденной результатами испытаний в соответствующей форме.
Примечание 2 - См. также 28.12, 28.14 и 28.15.
Примечание 3 - Требования по защите от изменений, вызванных различными видами лечения, для активных имплантируемых медицинских изделий особых видов могут быть уточнены в последующих частях ИСО 14708.
23 Защита активного имплантируемого медицинского изделия от воздействия механических сил
23.1 Части активного имплантируемого медицинского изделия, которые при нормальном использовании представляют собой ручное или портативное оборудование массой не более 10 кг, должны быть сконструированы таким образом, чтобы удар, вызванный падением или неправильным обращением при использовании, не повредил изделие.
Методика тестирования. Подвергают ручное или портативное оборудование активного имплантируемого медицинского изделия массой не более 10 кг испытанию на свободное падение в соответствии с МЭК 60068-2-32 при следующих условиях:
a) испытательная поверхность: твердое дерево плотностью не менее 630 кг/м и толщиной от 50 до 55 мм;
b) высота падения:
1) 1 м - для ручного оборудования,
2) 50 мм - для портативного оборудования;
c) положение, при котором падает образец, соответствует положению при нормальном использовании.
Соответствие считается подтвержденным, если после проведения испытания, описанного выше, испытавшая падение часть изделия функционирует согласно спецификации изготовителя для данной части изделия.
23.2 Имплантируемые или переносные пациентом части активного имплантируемого медицинского изделия, иные, нежели проводники или катетеры, должны быть сконструированы таким образом, чтобы выдерживать механические усилия, которые могут возникать при нормальных условиях использования.
Методика тестирования. Устанавливают каждую имплантируемую или переносную часть активного имплантируемого медицинского изделия в соответствии с рекомендациями МЭК 60068-2-47, приложение А, на испытательном оборудовании для оценки вибрационных воздействий (вибростенде) в соответствии с МЭК 60068-2-64 при следующих условиях:
a) частотный диапазон: от 5 до 150 Гц;
b) спектральный уровень ASD: 0,1 г/Гц;
c) продолжительность воздействия: 90 мин, из расчета 30 мин воздействий по каждому из трех взаимно перпендикулярных направлений;
d) воспроизводимость: средняя.
Соответствие считается подтвержденным, если активное имплантируемое медицинское изделие соответствует спецификации на изделие после проведения в полном объеме испытания, приведенного выше.
23.3 Имплантируемые проводники или катетеры должны выдерживать нагрузку растяжением, которая может возникнуть во время или после собственно имплантации, без разрушения какого-либо из проводников или образования трещин в электрической изоляции либо в теле проводника или катетера.
Соответствие проверяют анализом дизайна конструкции, представленного изготовителем и подтвержденного расчетами и результатами испытаний изготовителя в соответствующей форме.
23.4 Имплантируемые проводники, имеющие соединения двух и более проводящих компонентов, должны быть сконструированы таким образом, чтобы в области соединения не возникало изгибных напряжений от нагрузки, которая может иметь место при выполнении имплантации или после нее.
Соответствие проверяют анализом информации, предоставленной изготовителем и подтвержденной результатами испытаний от изготовителя в соответствующей форме.
23.5 Имплантируемые проводники или катетеры должны выдерживать изгибные напряжения, которые могут возникать при имплантации или после нее, без признаков механических разрушений какого-либо из проводников или образования трещин в обмотке электрической изоляции либо в теле проводника или катетера.
Соответствие подтверждают анализом информации, предоставленной изготовителем и подтвержденной результатами испытаний от изготовителя в соответствующей форме.
23.6 Имплантируемые коннекторы, предназначенные для использования медицинским персоналом с целью соединения имплантируемых изделий или их принадлежностей, должны быть идентифицированы (см. 8.2 и 9.2). Изготовитель должен утвердить их назначение при имплантации (см. 28.4) согласно следующей методике.
Методика тестирования. Погружают собранную пару коннекторов в солевой раствор концентрацией приблизительно 9 г/л при комнатной температуре не менее чем на 8 ч. После извлечения из солевого раствора эту же пару коннекторов подвергают воздействию разделяющей силы (5,0±0,5) Н, (7,5±0,5) Н и (10,0±0,5) Н каждый раз не менее чем в течение 10 с и относительного крутящего момента, равного (0,02±0,005) Н·м. Если собранная пара коннекторов сконструирована так, что допускает поворот частей пары относительно друг друга, то при испытании крутящий момент не надо прикладывать. Записывают значение максимальной силы, при которой не произошло разделение коннекторов в качестве результата испытания.
Соответствие подтверждают оценкой протоколов испытания изготовителя.
24 Защита активного имплантируемого медицинского изделия от повреждений, вызванных электростатическим разрядом
Неимплантируемые части активного медицинского изделия должны быть сконструированы таким образом, чтобы электростатический разряд, который может возникнуть при обращении с изделием, не вызвал в нем необратимых изменений.
Методика тестирования. Подключают к сети изделие согласно инструкции изготовителя. Неимплантируемая часть должна выдерживать воздействие электростатического разряда согласно описанию в МЭК 61000-4-2 (при условиях окружающей среды точно по указаниям 8.1.1), равного 2 кВ в случае контактного разряда на проводящей поверхности и 8 кВ в случае разряда в воздухе на изолированных поверхностях. К каждой испытуемой точке необходимо подвести не менее 10 раз разряд, равный 2 кВ, и не менее 5 раз - 8 кВ.
Соответствие считается подтвержденным, если активное имплантируемое медицинское изделие функционирует в безопасном режиме и может быть перезагружено для обеспечения всех функций, заявленных в спецификации изготовителя.
Примечание - Требования по защите от повреждения электростатическим разрядом для определенных видов активных имплантируемых медицинских изделий могут быть уточнены в последующих частях ИСО 14708.
25 Защита активного имплантируемого медицинского изделия от повреждений, вызванных изменениями атмосферного давления
Имплантируемые части активного имплантируемого медицинского изделия должны быть сконструированы таким образом, чтобы противостоять изменениям давления, которые могут возникнуть при транспортировании или нормальных условиях использования.
Соответствие подтверждают анализом дизайна конструкции и оценкой отчета изготовителя о результатах испытаний по оценке последствий деформации изделия при значениях давления (70±3,5) кПа и (150,0±7,5) кПа, прикладываемого в течение не менее 1 ч.
Примечание - Требования по защите от повреждения, вызванного изменением атмосферного давления, для активных имплантируемых медицинских изделий определенных видов могут быть уточнены в последующих частях ИСО 14708.
26 Защита активного имплантируемого медицинского изделия от повреждений, вызванных перепадами температур
26.1 Неимплантируемые части активного имплантируемого медицинского изделия, работающие от источников электроэнергии, должны быть разработаны и изготовлены таким образом, чтобы соответствовать требованиям МЭК 60601-1, раздел 42.
Соответствие подтверждается в соответствии с требованиями МЭК 60601-1.
26.2 Имплантируемые части активного имплантируемого медицинского изделия должны быть разработаны и изготовлены таким образом, чтобы изменения температуры, которые могут возникать при хранении или транспортировании, не вызвали в них необратимых изменений.
Методика тестирования. Имплантируемые части активного имплантируемого медицинского изделия, находящиеся только в стерильной упаковке, подвергают испытанию согласно МЭК 60068-2-14 (см. испытание Nb) при следующих условиях:
a) при низкой температуре: самом нижнем значении температуры хранения, установленной изготовителем, или при минус (10±3) °С (смотря, что выше);
b) при высокой температуре: самом верхнем значении температуры хранения, установленной изготовителем, или при (55±2) °С (смотря, что ниже);
c) при скорости изменения температуры (1,0±0,2) °С/мин.
Соответствие считается подтвержденным, если активное имплантируемое медицинское изделие соответствует спецификации на изделие после проведения испытания. Если температуры при тестировании иные, нежели минус (10±3) °C и (55±2) °C, то это должно быть отражено в протоколе испытаний.
27 Защита активного имплантируемого медицинского изделия от электромагнитного неионизирующего излучения
Имплантируемые части активного имплантируемого медицинского изделия не должны вызывать какого-либо вреда, обусловленного чувствительностью к электромагнитному воздействию, связанному с внешними электромагнитными полями, ни за счет неправильного функционирования изделия, ни за счет повреждения изделия, ни за счет нагрева изделия, ни за счет вызванного локального увеличения плотности индуцированного электрического тока у пациента.
Методика испытания. Определяют возможные опасности, принимая во внимание электромагнитное поле в окружающей среде, в которой предусмотрено применение активного имплантируемого медицинского изделия. Для каждого потенциального источника вреда оценивают вероятность вреда путем анализа дизайна конструкции с точки зрения контроля опасности любого вида. При необходимости подтверждают анализ дизайна результатами испытаний.
Примечание 1 - В первом приближении принимают значение магнитной интенсивности равным 150 А/м при изменении частоты от 100 кГц до максимального значения 30 МГц. Электрическое поле исследовать не нужно.
Соответствие подтверждают экспертизой соответствующей документации, подготовленной изготовителем.
Примечание 2 - Требования по защите активных имплантируемых медицинских изделий определенных видов от электромагнитного ионизирующего излучения могут быть уточнены в последующих частях ИСО 14708.
28 Сопровождающая документация
28.1 Сопровождающая документация должна содержать наименование и адрес изготовителя, включая страну и город.
Соответствие проверяют в ходе осмотра.
28.2 Если упаковка содержит какие-либо радиоактивные вещества, то сопровождающая документация должна содержать информацию, указывающую тип и степень активности радиоактивных веществ (см. также раздел 18).
Соответствие проверяют в ходе осмотра.
28.3 Сопровождающая документация должна содержать описание изделия (например, кардиостимулятор) и обозначение изделия.
Соответствие проверяют в ходе осмотра.
28.4 Если упаковка содержит имплантируемую часть активного имплантируемого медицинского изделия, предусмотренную для соединения с другим имплантируемым изделием или имплантируемой принадлежностью, в сопровождающую документацию должны быть включены данные о максимальном значении усилия фиксации соединения, полученном согласно 23.6.
Соответствие проверяют в ходе осмотра.
28.5 В сопровождающей документации должна быть включена информация - перечень принадлежностей, которые могут потребоваться для работы с изделием и для его основных функций.
Соответствие проверяют в ходе осмотра.
28.6 В сопровождающей документации должно содержаться пояснение метода интерпретации идентификационных кодов, требуемых 13.3.
Соответствие проверяют в ходе осмотра.
28.7 Если применимо, в сопровождающую документацию должна быть включена информация, относящаяся к лекарственным средствам, доставку которых предполагается осуществлять, используя активное имплантируемое медицинское изделие (см. также 14.4 и 19.5).
Примечание - Этот пункт неприменим к лекарственным средствам, являющимся неотъемлемой частью активного имплантируемого медицинского изделия.
Соответствие проверяют в ходе осмотра.
28.8 В сопровождающую документацию должно быть включено описание предполагаемого применения изделия, даны спецификации на изделие и представлена информация о значимых побочных эффектах (см. 19.4).
Соответствие проверяют в ходе осмотра.
28.9 В сопровождающую документацию должна быть включена информация, позволяющая терапевту выбрать подходящее изделие, принадлежности и связанные с ним изделия (например, программатор).
Соответствие проверяют в ходе осмотра.
28.10 В сопровождающую документацию должна быть включена инструкция по применению активного имплантируемого медицинского изделия, чтобы врач и, если допустимо, пациент могли использовать его правильно.
Соответствие проверяют в ходе осмотра.
28.11 В сопровождающую документацию должна быть включена информация об опасностях, которых можно избежать при имплантации.
Соответствие проверяют в ходе осмотра.
28.12 В сопровождающую документацию должны быть включены предупреждения, касающиеся использования медицинского изделия, включая информацию об опасностях, которые могут быть вызваны взаимодействием имплантируемого изделия и другого оборудования, которое могут применять в других клинических процедурах, или медицинских манипуляциях, например таких, как упомянуты в 20.2 и разделах 21, 22 и 27.
Соответствие проверяют в ходе осмотра.
28.13 В сопровождающую документацию должны быть включены предупреждения о том, что если к пациенту с активным имплантируемым медицинским изделием применяется какое-либо лечебное воздействие, при котором через его тело проходит электрический ток, формируемый внешним источником, то изделие должно быть сначала дезактивировано либо следует уделить особое внимание мониторингу рабочих характеристик активного имплантируемого медицинского изделия на начальной стадии лечения.
Соответствие проверяют в ходе осмотра.
28.14 В сопровождающую документацию должны быть включены предупреждения о том, что на имплантируемые части активного имплантируемого медицинского изделия не должно оказываться воздействия ультразвуком терапевтического уровня, поскольку изделие может неадекватно сконцентрировать ультразвуковое поле и причинить вред.
Соответствие проверяют в ходе осмотра.
28.15 В сопровождающую документацию должно быть включено предупреждение о том, что электрические компоненты в активном имплантируемом медицинском изделии могут быть повреждены терапевтическим ионизирующим излучением и что повреждение изделия, возможно, не будет выявлено сразу.
Соответствие проверяют в ходе осмотра.
28.16 В сопровождающую документацию должно быть включено заявление о том, что имплантируемые части активного имплантируемого медицинского изделия стерильны.
Соответствие проверяют в ходе осмотра (см. также раздел 11).
28.17 Если применимо, в сопровождающую документацию должны быть включены инструкции по методу стерилизации принадлежностей, поставляемых нестерильными, и по работе с содержимым стерильной упаковки в случае, если она была повреждена или вскрывалась ранее.
Соответствие проверяют в ходе осмотра.
28.18 Если применимо, сопровождающая документация должна содержать предупреждение о том, что имплантируемые части недопустимо использовать повторно, если до этого они были имплантированы другому пациенту. В противном случае сопровождающая документация, поставляемая с имплантируемыми частями, должна содержать предупреждение о том, что изделие можно использовать повторно, только если оно было восстановлено под контролем изготовителя.
Соответствие проверяют в ходе осмотра.
28.19 Если изделие снабжено источником питания, то сопровождающая документация должна содержать информацию, позволяющую установить срок службы изделия в случае, если изделие используется при нормальных настройках, установленных изготовителем, и в случае, если в параметры функционирования были внесены изменения, в результате которых наблюдались максимальные утечки тока, соответствующие по величине требованиям клинического применения.
Соответствие проверяют в ходе осмотра.
28.20 В сопровождающую документацию должно быть включено предупреждение о рекомендуемых мерах предосторожности для предотвращения неблагоприятных эффектов, связанных с изменением рабочих характеристик активного имплантируемого медицинского изделия.
Соответствие проверяют в ходе осмотра.
28.21 В сопровождающую документацию должна быть включена информация о специальных условиях окружающей среды или ограничениях по транспортированию (например, о предотвращении ударных воздействий, вибрации, температуре, давлении и влажности), необходимая для правильного транспортирования и хранения изделия.
Соответствие проверяют в ходе осмотра.
28.22 В сопровождающую документацию должны быть включены меры предосторожности, которые необходимо предпринять для предотвращения неблагоприятного воздействия на пациента в связи со специфическими неблагоприятными условиями окружающей среды (например, наличием электромагнитных помех, экстремальных температур, скачков давления).
Соответствие проверяют в ходе осмотра.
28.23 В сопровождающую документацию должны быть включены советы о том, что пациенту следует уточнить медицинские рекомендации, перед тем как попасть в окружающую среду, условия которой могут неблагоприятно отразиться на работе активного имплантируемого медицинского изделия, в том числе области, защищенные надписями о недопустимости входа пациентов с кардиостимуляторами.
Соответствие проверяют в ходе осмотра.
28.24 Если применимо, в сопровождающую документацию должна быть включена инструкция о приемлемом способе удаления или извлечения активного имплантируемого медицинского изделия.
Соответствие проверяют в ходе осмотра.
Приложение A
(справочное)
Соответствие между фундаментальными принципами ИСО/ТО 14283 и разделами настоящего стандарта
Таблица A.1
Фундаментальные принципы ИСО/ТО 14283 | Разделы настоящего стандарта и охватываемые аспекты |
3 Общие принципы | 8.1 Требуется, чтобы предупреждающие надписи были заметными |
3.2 Решения, принятые изготовителем для разработки и конструирования имплантатов, должны соответствовать принципам безопасности с учетом общего уровня знаний и технического развития. При выборе наиболее подходящего решения изготовитель должен руководствоваться перечисленными ниже принципами в следующем порядке: | (Настоящий принцип является фундаментальным по отношению ко всем активным имплантируемым медицинским изделиям, для которых предназначен ИСО 14708. Данный подход в основном относится к содержанию требований разделов 14, 19 и 21) |
3.3 Имплантаты должны достигать рабочих характеристик, предусмотренных изготовителем, и должны быть разработаны, изготовлены и упакованы таким образом, чтобы они были пригодны для выполнения одной или более функций, приведенных в 2.1 (ИСО/ТО 14283), как указано изготовителем | 10.4 Требуется, чтобы сопровождающая информация была нанесена на изделие |
3.4 На характеристики и изделия, приведенные в 3.1, 3.2 и 3.3 (ИСО/ТО 14283), не должно быть негативного влияния в такой степени, чтобы нарушались клинические условия и безопасность пациентов и, где применимо, других лиц, в течение срока служба имплантата, указанного изготовителем, при воздействии на имплантат нагрузок, возникающих при нормальном использовании имплантата | 19.2 Требуется наличие индикатора истощения источника энергии. |
3.5 Имплантаты должны быть разработаны, изготовлены и упакованы таким образом, чтобы на их характеристиках и работе в течение периода предполагаемого использования отрицательно не сказывались транспортирование и хранение, принимая во внимание инструкции и информацию, указанную изготовителем | 7.2 Требуется, чтобы стерильная упаковка была защищена товарной упаковкой. |
3.6 В отношении любого нежелательного побочного эффекта необходимо оценить приемлемость риска относительно предполагаемых рабочих характеристик | 19.3 Требуется определить методику подтверждения того, что условия единичного отказа не вызывают причинения вреда. |
4 Специальные принципы, касающиеся дизайна конструкции | |
a) выбору используемых материалов, в частности оценке токсичности и, где применимо, свойствах возгораемости; | 14.3 Требуется провести оценку биосовместимости. |
b) совместимости используемых материалов и биологических тканей, клеток и тканевых жидкостей, принимая во внимание предполагаемое назначение имплантата. | 14.3 Требуется провести оценку биосовместимости. |
4.1.2 Имплантаты должны быть разработаны, изготовлены и упакованы таким образом, чтобы минимизировать риск, вызванный загрязнением и наличием остаточных веществ, для лиц, занятых транспортированием, хранением и использованием имплантатов, и пациентов, принимая во внимание предполагаемое назначение изделия. Особое внимание необходимо уделить незащищенным тканям и продолжительности и частоте воздействия. | 14.2 Требуется провести оценку на загрязнение частицами. |
4.1.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы они могли быть использованы безопасно с материалами, веществами и газами, с которыми они контактируют при нормальном использовании или при рутинных процедурах; если предполагается использование имплантатов для доставки лекарственных веществ, то они должны быть разработаны и изготовлены таким образом, чтобы быть совместимыми с лекарственным веществом в соответствии с положениями и ограничениями для этих веществ и чтобы их рабочие характеристики поддерживались в соответствии с предполагаемым использованием. | 19.5 Требуется доказать совместимость с лекарственными веществами. |
4.1.4 В случае если в имплантаты включено как неотъемлемая часть вещество, которое, если применяется отдельно, может быть рассмотрено как лекарственное вещество согласно определению 2.4 (ИСО/ТО 14283) и которое может воздействовать на организм дополнительно к влиянию, оказываемому изделием, его безопасность должна быть подтверждена, принимая во внимание предполагаемое использование имплантированного изделия. | 14.4 Требуется оценить качество и безопасность содержащихся в имплантате лекарственных веществ. |
4.1.5 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы свести к минимуму риск, вызванный утечкой вещества из имплантата. | 25 Требуется, чтобы имплантируемые части выдерживали изменения давления. |
4.1.6 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы уменьшить, насколько возможно, риски, вызванные случайным проникновением вещества в имплантат, принимая во внимание имплантат и характер окружающей среды, в которой предполагается его использовать. | 25 Требуется, чтобы имплантируемые части выдерживали изменения давления. |
4.1.7 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски пациента или пользователя системы, включая программное обеспечение | 19.3 Требуется провести анализ дизайна разработки и выбрать методику анализа |
4.2 Инфекция и микробное заражение | 14.1 Требуется, чтобы изделие поставлялось стерильным. |
4.2.2 Ткани животного происхождения должны быть поставлены от животных, подвергнутых ветеринарным контролю и надзору, адаптированным к предполагаемому применению ткани. | Не применимо к активным имплантируемым медицинским изделиям |
4.2.3 Имплантаты, поставляемые стерильными, должны быть разработаны, изготовлены и упакованы в защитную упаковку, которая создает антимикробный барьер, для обеспечения их стерильности при поступлении на рынок и при условиях хранения и транспортирования, оговоренных изготовителем, до тех пор, пока упаковка не будет вскрыта или повреждена. | 7.1 Требуется поставка изделия в одноразовой упаковке. |
4.2.4 Имплантаты, поставляемые стерильными, должны быть изготовлены и стерилизованы, если применимо, валидированным методом. | 14.1 Требуется подтверждать, что изделие стерилизовалось с использованием валидированного процесса. |
4.2.5 Имплантаты, для которых предполагается стерилизация, должны быть изготовлены в соответствующих контролируемых условиях (например, окружающей среды). | 14.1 Требуется поставка изделия в стерильном виде. |
4.2.6 Система упаковки нестерильных имплантатов должна поддерживать на оговоренном уровне чистоты изделие без ухудшения и, если имплантаты стерилизуют перед использованием, минимизировать риск микробного заражения; система упаковки должна соответствовать методу стерилизации, указанному изготовителем. | Не применимо к активным имплантируемым медицинским изделиям. |
4.2.7 Упаковка и/или этикетка имплантата должны устанавливать различие между идентичными или похожими изделиями в стерильном и нестерильном состояниях | Не применимо к активным имплантируемым медицинским изделиям |
4.3 Свойства конструкции и окружающей среды | |
4.3.1 Если имплантат предназначен для использования совместно с другим изделием или оборудованием, вся комбинация, включая систему соединений, должна быть безопасной и не должна ухудшать специфических свойств изделия. Любые ограничения по использованию должны быть указаны на этикетке или в инструкции по применению. | 9.9 Требуется, чтобы соединения имплантируемого изделия были идентифицированы. |
4.3.2 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы устранить или минимизировать, насколько это возможно: | |
a) риск травмы с учетом их физических особенностей, таких как соотношение объем/давление, размерами и, если применимо, эргономическими особенностями; | 15.1 Требуется установить требования к качеству поверхностей неимплантируемых частей. |
b) риски, связанные с разумными, предсказуемыми условиями окружающей среды, такими как магнитные поля, воздействиями внешнего электричества, электростатического разряда, давления, температуры или изменений давления и ускорения; | 23.1 Требуется провести испытание неимплантируемых частей на падение. |
c) риски обоюдных помех при использовании с другими изделиями (такими как дефибрилляторы или высокочастотные хирургические инструменты), обычно используемыми для исследований или при данном виде лечения; | 20.1 Требуется защита внешних электродов ЭКГ от дефибрилляции. |
d) риски, которые могут возникнуть при условии, что обслуживание и калибровка невозможны, включая (если применимо): чрезмерное увеличение тока утечки, старение материалов, усиление нагрева, генерируемого имплантатом, снижение точности любого измерительного или контрольного механизма. | 17 Требуется оценить воздействие локального нагрева, вызванного отказом имплантируемого изделия. |
4.3.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски возгорания или взрыва при нормальных условиях эксплуатации или в случае единичного отказа. Риски "при нормальных условиях или в случае единичного отказа" означают риски, которые можно оценить путем анализа рисков. Особое внимание следует обратить на имплантаты, предполагаемое использование которых включает воздействие огнеопасных веществ или веществ, которые могут вызвать возгорание | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия |
4.4 Имплантаты с функцией измерения | |
4.4.1 Имплантаты с функцией измерения должны быть разработаны и изготовлены таким образом, чтобы обеспечить установленную точность и стабильность в заданных пределах точности с учетом предполагаемого использования имплантата. Пределы точности должны быть установлены изготовителем. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.4.1.1 Методы выполнения измерений с помощью шкалы на дисплее или в режиме наблюдения должны быть разработаны в соответствии с принципами эргономики, с учетом предполагаемого использования имплантата. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.4.1.2 Если на имплантате или его принадлежностях приведены инструкции по работе с имплантатом или работе индикации, или параметрам настройки с помощью визуальной системы, эта информация должна быть понятна пользователю и, если необходимо, пациенту. | 13.4 Требования к индикаторам визуального типа. |
4.4.2 Измерения, осуществляемые с помощью имплантатов, снабжённых функцией измерения, должны быть выражены в единицах, соответствующих положениям серии ИСО 31 | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия |
4.5 Защита от излучения | |
4.5.1 Общие положения | (См. более подробную информацию ниже) |
4.5.2 Предусмотренное излучение | В настоящее время не применимо к активным имплантируемым медицинским изделиям. |
4.5.3 Непредусмотренное излучение | 9.1 Требуется использовать маркировку с предупреждениями о любых радиоактивных веществах. |
4.5.4 Инструкции | В настоящее время не применимо к активным имплантируемым медицинским изделиям |
4.6 Ионизирующее излучение | В настоящее время не применимо к активным имплантируемым медицинским изделиям |
4.7 Принципы, касающиеся имплантатов, подсоединенных или оборудованных источником энергии | |
4.7.1 Имплантаты со встроенными электронными программируемыми системами должны быть разработаны таким образом, чтобы обеспечивать воспроизводимость, надежность и эффективность этих систем в соответствии с предполагаемым использованием. При возникновении рисков (рисков системы), установленных при анализе рисков для данного изделия/системы, должны быть приняты соответствующие меры по уменьшению или устранению риска, насколько возможно. | 19.3 Требуется анализ дизайна конструкции и выбор метода анализа. |
4.7.2 Имплантаты, в которых безопасность пациента зависит от внутреннего источника энергии, должны быть оснащены средствами индикации состояния источника питания. | 19.2 Требуется наличие индикатора истощения источника питания. |
4.7.3 На имплантатах, насколько возможно, должен быть нанесен код, с помощью которого тип имплантата или изготовители могут быть идентифицированы (в частности, в соответствии с типом имплантата); при необходимости код должен быть читаем без хирургического вмешательства. | 13.3 Требование установлено и расширено. |
4.7.4 Для имплантатов, в которых безопасность пациента зависит от внешнего источника энергии, внешний источник энергии должен включать в себя систему сигнализации для подачи сигнала в случае отказа питания. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.5 Внешние изделия, предназначенные для мониторинга одного или более клинических параметров имплантата, должны быть оснащены системой сигнализации для подачи сигнала пользователю в ситуации, которая может привести к смерти или серьезному ухудшению состояния пациента. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.6 Защита от электрических рисков | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.6.2 Активные имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, связанные с использованием источников энергии, в первую очередь при использовании электроэнергии, уделяя особое внимание изоляции, токам утечки и перегреву изделия. | 16.2 Требуется установить безопасные пределы утечки тока от имплантируемых частей. |
4.7.7 Защита от механических рисков | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.7.2 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, возникающие от вибрации, генерируемой изделием, принимая во внимание технический прогресс и средства, доступные для снижения вибрации, особенно источника энергии, если только вибрация не является частью специальных рабочих характеристик изделий. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.7.3 Имплантаты должны быть разработаны и изготовлены таким образом, чтобы минимизировать риски, возникающие из-за шумов, принимая во внимание технический прогресс и средства, доступные для снижения шумов, особенно от источника энергии, если только шумы не являются частью специальных рабочих характеристик изделий. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.7.4 Терминалы и коннекторы для источников электрической, газовой или гидравлической и пневматической энергии, с которыми приходится работать пользователю, должны быть разработаны и сконструированы таким образом, чтобы минимизировать все возможные риски. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.8.1 Имплантаты должны быть разработаны и сконструированы таким образом, чтобы собственно функционирование программируемых и контрольных систем, включая программное обеспечение, не ухудшало безопасность пациента и пользователя, принимая во внимание предусмотренные назначением изделия условия применения. | 19.3 Требуется анализ дизайна конструкции и выбор методики этого анализа. |
4.7.8.2 Имплантаты, разработанные для подачи энергии или введения лекарственных веществ, должны быть разработаны и сконструированы таким образом, чтобы параметры потока могли устанавливаться и поддерживаться достаточно точно для минимизации риска для пациента. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.8.3 Имплантаты, разработанные для введения лекарственных веществ, должны включать в себя соответствующие средства предотвращения и/или индикации любой некорректной скорости потока, которая может привести к опасности. | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия. |
4.7.8.4 Имплантаты, разработанные для подачи энергии или введения лекарственных веществ, должны быть разработаны и сконструированы со встроенными средствами минимизации рисков случайного превышения опасного уровня энергии или лекарственного вещества | 5 Требуется применять МЭК 60601-1 к неимплантируемым частям активного имплантируемого медицинского изделия |
4.8 Информация, предоставляемая изготовителем | |
4.8.1 Каждый имплантат должен поставляться с сопровождающей информацией, необходимой для его безопасного использования и идентификации изготовителя, с учетом уровня подготовки и знаний потенциального пользователя. | 10.4 Требуется, чтобы сопровождающая документация была нанесена на изделие. |
4.8.2 Если применимо, информацию следует представлять в виде символов. Любой символ или цветовая индикация должны соответствовать международным стандартам. Если стандарта не существует, символ должен быть описан в документации, поставляемой с имплантатом. | 4 Разрешается использование символов, аббревиатур и цветовой индикации. |
4.8.3 Этикетка должна содержать следующие сведения: | 5 Требуется соблюдение требований к этикеткам МЭК 60601-1 для неимплантируемых частей. |
b) подробные сведения, необходимые пользователю для идентификации имплантата и содержимого упаковки; | 9.3 Требуется поместить описание изделия и обозначение модели на товарной упаковке. |
c) если применимо, индикацию, что содержимое упаковки стерильно (например, "СТЕРИЛЬНО"); | 9.5 Требуется нанесение утверждения, что упаковка была простерилизована. |
d) если применимо, номер партии или серийный номер, которому предшествует соответствующая идентификация (например, "LOT" или "SN"); | 9.3 Требуется поместить описание изделия и обозначение модели на товарной упаковке. |
e) если применимо, дата, до которой имплантат может быть использован; | 9.7 Требуется нанесение маркировки "использовать до". |
f) индикацию, что имплантаты для одноразового использования; | 28.18 Требуется выбрать вид предупреждающей надписи о повторном использовании изделия. |
g) если применимо, указание на специальное назначение (например, "изделие, сделанное на заказ" или "только для клинических исследований"); | 9.12 Требуется нанесение маркировки специального назначения. |
h) любые специальные условия хранения и/или транспортирования; | 9.11 Требуется нанесение маркировки с информацией о любых специальных условиях окружающей среды или ограничениях по транспортированию. |
i) любые специальные рабочие инструкции; | Для имплантируемых частей активного имплантируемого медицинского изделия все рабочие инструкции приводятся в сопровождающей документации. |
j) любые предупреждения или меры предосторожности, которые следует предпринять; | В общем случае предупреждения и меры предосторожности, за исключением тех, что связаны со специальными условиями транспортирования [см. 4.8.3, перечисление h)], должны быть описаны в сопровождающей документации, а не на этикетке. |
k) для активных имплантатов - месяц и год производства; | 9.6 Требуется нанесение маркировки с выбором формата. |
l) если применимо, метод стерилизации. | 11.2 Требуется, чтобы на маркировке был указан метод стерилизации. |
4.8.4 Если предполагаемое использование имплантата не очевидно для пользователя, изготовитель должен четко указать назначение изделия на этикетке и в инструкции по применению. | 9.10 Требуется дополнительное описание, если требования 9.3 и 9.4 не соблюдены в полной мере. |
4.8.5 Если это разумно и применимо, имплантаты и съемные компоненты должны быть идентифицированы по возможности в терминах серийного номера или номера партии для обеспечения возможности проведения всех надлежащих действий после установления любых потенциальных рисков, обусловленных применением как имплантата, так и съемных компонентов. | 8.2 Требуется отслеживать имплантируемые части. |
4.8.6 Если применимо, инструкция по применению должна содержать следующие сведения: | |
а) детали, упомянутые в 4.8.3, за исключением перечислений d), e) и i); | 28.1 Требуется указать наименование и адрес изготовителя. |
b) рабочие характеристики, приведенные в ИСО/ТО 14283, подраздел 3.3, и любые нежелательные побочные эффекты; | 28.8 Требуется поместить информацию о предполагаемом использовании и характеристиках, а также о возможных побочных эффектах. |
c) если имплантат используется в сочетании с другими медицинскими изделиями или оборудованием, как требует его предполагаемое использование, необходимые подробности о его характеристиках для определения корректных используемых имплантатов или оборудования для получения безопасной комбинации; | 28.4 Требуется указать сведения о максимальной достоверной величине усилия фиксации соединения. |
d) всю информацию, необходимую для подтверждения того, что имплантат используется надлежащим образом и может работать правильно и безопасно, а также, где применимо, сведения, позволяющие оценить срок службы источника энергии; | 28.10 Требуется предоставить полную инструкцию по применению. |
e) если применимо, информацию, необходимую для предотвращения специальных рисков, связанных с имплантацией имплантата; | 28.11 Требуется предоставить сведения по предотвращению опасностей во время имплантации. |
f) информацию, относящуюся к рискам перекрестного влияния, вызванного присутствием имплантата во время проведения специфического лечения или обследований; | 28.12 Требуется нанести предупреждающие надписи об опасностях, возникающих при взаимодействии. |
g) необходимые инструкции на случай повреждения стерильной упаковки и, если применимо, подробные сведения о допустимом методе повторной стерилизации; | 28.17 Требуется указать меры предосторожности при работе со вскрытой или поврежденной стерильной упаковкой. |
h) если имплантаты поставляются с условием, что они будут стерилизованы перед использованием, инструкция по очистке и стерилизации должна быть такой, что при ее соблюдении имплантат будет соответствовать требованиям ИСО/ТО 14283, раздел 3; | 28.17 Требуется указать в инструкции особенности стерилизации принадлежностей, поставляемых нестерильными. |
i) детали последующей обработки или обслуживания, требуемые до использования имплантата (например, стерилизация, окончательная сборка и т.д.); | Неприменимо, т.к. в 14.1 требуется, чтобы активные имплантируемые медицинские изделия поставлялись стерильными. |
j) в случае если имплантат испускает радиоактивное излучение с лечебными целями, сведения о характере, типе, интенсивности и распространении этого излучения. | Неприменимо к активным имплантируемым медицинским изделиям. |
k) меры предосторожности, соблюдаемые в случае изменения рабочих характеристик имплантата; | 28.19 Требуется указать сведения, позволяющие оценить срок службы источника энергии. |
l) меры предосторожности в ответ на воздействие в разумно предсказуемых условиях окружающей среды, например, магнитного поля, внешних источников электроэнергии, электростатических разрядов, давления или перепада давлений, ускорения, источников тепла и т.д.; | 28.22 Требуется указать сведения по мерам предосторожности, которые необходимо предпринять, чтобы защититься от негативного влияния окружающей среды. |
m) достаточный объем информации, касающийся лекарственных веществ или веществ, которые должны доставляться с помощью данного имплантата, включая любые ограничения при выборе веществ, которые необходимо доставлять; | 28.25 Требуется указать сведения о надлежащем извлечении имплантата |
4.9 Клиническая оценка | |
a) сбора данных из соответствующей научной литературы, доступной в настоящее время, по назначению изделия, предусмотренного изготовителем, либо | 19.4 Требуется оценить нежелательные воздействия, вызванные изделием. |
b) результатов всех клинических исследований, проводимых таким образом, чтобы защищать пациентов и обеспечивать при этом научный характер исследования | 19.4 Требуется оценить нежелательные воздействия, вызванные изделием. |
Приложение B
(справочное)
Связь между разделами настоящего стандарта и фундаментальными принципами, перечисленными в приложении A
Таблица B.1
Подраздел | Соответствующий фундаментальный принцип |
4 | 4.8.2 |
5 | 4.4.1, 4.4.1.1, 4.4.1.2, 4.7.4, 4.7.5, 4.7.7.1, 4.7.7.2, 4.7.7.3, 4.7.7.4, 4.7.8.2, 4.7.8.3, 4.7.8.4, 4.8.3 и 4.8.6, перечисление p) |
7.1 | 4.2.3 |
7.2 | 3.5 и 4.2.3 |
8.1 | 3.1 |
8.2 | 4.8.5 |
9.1 | 4.5.3 |
9.2 | 4.8.3, перечисление a) |
9.3 | 4.8.3, перечисление b), и 4.8.3, перечисление d) |
9.4 | 4.8.3, перечисление b) |
9.5 | 4.8.3, перечисление c) |
9.6 | 4.8.3, перечисление k) |
9.7 | 4.8.3, перечисление e) |
9.8 | 4.8.3, перечисление b) |
9.9 | 4.3.1 |
9.10 | 4.8.3, перечисление b), и 4.8.4 |
9.11 | 4.8.3, перечисление h) |
9.12 | 4.8.3, перечисление g) |
10.1 | 3.5 и 4.2.3 |
10.2 | 3.5 и 4.2.3 |
10.3 | 3.5 |
10.4 | 3.3 и 4.8.1 |
11.1 | 4.8.3, перечисление a) |
11.2 | 4.8.3, перечисление c), и 4.8.3, перечисление l) |
11.3 | 4.8.3, перечисление c) |
11.4 | 4.8.3, перечисление k) |
11.5 | 4.8.3, перечисление e) |
11.6 | 4.8.3, перечисление b), и 4.8.3, перечисление e) |
11.7 | 4.8.3, перечисление b), и 4.2.3 |
11.8 | 4.3.1 |
11.9 | 4.2.3 |
11.10 | 4.8.3, перечисление g) |
12.1 | 4.2.3 |
12.2 | 4.2.3 |
12.3 | 3.5 |
13.1 | 4.8.5 |
13.2 | 4.8.5 |
13.3 | 4.7.3 |
13.4 | 4.4.1.2 |
14.1 | 4.2.1, 4.2.3, 4.2.4 и 4.2.5 |
14.2 | 4.1.2 и 4.2.5 |
14.3 | 4.1.1, перечисление a), 4.1.1, перечисление b), и 4.1.2 |
14.4 | 4.1.4 |
15.1 | 4.3.2, перечисление a) |
15.2 | 4.3.2, перечисление a) |
16.1 | 4.7.6.1 |
16.2 | 4.7.6.2 |
16.3 | 4.7.6.2 |
17 | 4.7.6.2 и 4.3.2, перечисление d) |
18.1 | 4.5.3 |
18.2 | 4.5.3 |
18.3 | 4.5.3 |
19.1 | 4.3.2, перечисление d) |
19.2 | 4.3.2, перечисление d) |
19.3 | 4, 4.3.2, перечисление d), и 4.7.2 |
19.4 | 4, 3.6, 4.1.7, 4.7 и 4.7.8.2 |
19.5 | 4.1.3 |
20.1 | 4.3.2, перечисление c) |
20.2 | 4.3.2, перечисление c) |
21 | 4.3.2, перечисление c) |
23.1 | 3.4 и 4.3.2, перечисление b) |
23.2 | 3.4 и 4.3.2, перечисление b) |
23.3 | 3.4 |
23.4 | 3.4 |
23.5 | 3.4 |
23.6 | 3.4 и 4.3.1 |
24 | 4.3.2, перечисление b) |
25 | 4.3.2, перечисление b) |
26.1 | 3.4 и 4.7.6.2 |
26.2 | 3.5 и 4.3.2, перечисление b) |
27 | 4.3.2, перечисление b) |
28.1 | 4.8.6, перечисление а), 4.8.3, перечисление a) |
28.2 | 4.5.3 |
28.3 | 4.8.6 перечисление а), 4.8.3, перечисление b) |
28.4 | 3.4, 4.3.1 и 4.8.6, перечисление c) |
28.5 | 4.3.1 и 4.8.6, перечисление c) |
28.6 | 4.7.3 |
28.7 | 4.8.6, перечисление m) |
28.8 | 4.8.6, перечисления b) и o) |
28.9 | 4.8.6, перечисление c) |
28.10 | 4.8.6, перечисление d) |
28.11 | 4.8.6, перечисление e) |
28.12 | 4.3.2, перечисление c), и 4.8.6, перечисление f) |
28.13 | 4.3.2, перечисление c) |
28.14 | 4.3.2, перечисление c) |
28.15 | 4.3.2, перечисление c) |
28.16 | 4.8.6, перечисление a), 3.8.3, перечисление c) |
28.17 | 4.8.6, перечисление g) и 4.8.6, перечисление h) |
28.18 | 4.8.6, перечисление a), 4.8.3, перечисление f) |
28.19 | 4.8.6, перечисление k) |
28.20 | 4.8.6, перечисление k) |
28.21 | 4.8.6, перечисление a), 4.8.3, перечисление h) |
28.22 | 4.8.6, перечисление l) |
28.23 | 3.4 |
28.25 | 4.8.6, перечисление n) |
Приложение С
(справочное)
Обоснование
C.1 Введение
В ИСО/ТО 14283 приведен свод фундаментальных принципов, применяемых при разработке конструктивного решения и изготовлении хирургических имплантатов. Настоящий стандарт интерпретирует в целом эти фундаментальные принципы для активных имплантируемых медицинских изделий. Во многих пунктах настоящего стандарта интерпретация выполнена за счет уточнения деталей частных аспектов фундаментальных принципов и определения процедур оценки или испытаний.
Эти фундаментальные принципы составляют основу настоящего стандарта и последующих частей ИСО 14708. Их следует применять совместно с информацией, изложенной ниже. Для удобства фундаментальные принципы перечислены применительно к специальным разделам настоящего стандарта в приложении A.
C.2 Общие положения
Изложенные ниже замечания к некоторым положениям настоящего стандарта призваны облегчить его понимание. Настоящее приложение адресовано тем, кто знаком с устройством изделий или имеет опыт применения активных имплантируемых медицинских изделий, но не принимал участия в разработке настоящего стандарта. Содержание настоящего приложения относится не ко всем разделам настоящего стандарта; именно поэтому нумерация параграфов в приложении не является последовательной.
За исключением разделов 5, 7 и 8, разделы настоящего стандарта упорядочены, поэтому их можно рассматривать последовательно, начиная с проверки маркировки на внешней стороне товарной упаковки, затем проверки конструкции упаковки и далее вплоть до испытаний изделия, завершая все в итоге проверкой сопровождающей документации.
Для некоторых рисков в настоящем стандарте приведены специальные требования вместе с мерами оценки соответствия (например, оценки уровня тока утечки), которые в случае их удовлетворения адресуются одному или сразу нескольким фундаментальным принципам. Для других рисков настоящий стандарт требует, чтобы потенциальная опасность была идентифицирована и адресована путем использования процедуры менеджмента риска в соответствии с инструкциями, положенными в основу фундаментального принципа 3.2 ИСО/ТО 14283. Соответствие затем устанавливают проверкой документации, предоставленной изготовителем.
В некоторых случаях никакое лабораторное испытание ограниченной продолжительности не может обеспечить достаточную уверенность в характеристиках конкретного изделия или подтвердить рабочие характеристики изделия спустя несколько лет после имплантации. В таких случаях ИСО 14708 требует от изготовителя изделия подготовить документированные исследования, необходимые для экспертного уровня оценки изделия.
По своей природе общий стандарт не может быть применен в одинаковой степени ко всем изделиям, относящимся к его области применения. Например, менеджмент риска может показать, что конкретное изделие требует более жестких пределов по конкретному параметру (например, по уровню тока утечки при использовании весьма малых размеров электродов). В другом случае границы температурного диапазона могут быть менее жесткими, если доступные технологии способствуют большему уровню нагрева, но их применение позволяет теплоте безопасно рассеяться.
Возможно, по какому-либо особому виду активных имплантируемых изделий требуется уделять особое внимание при применении настоящего стандарта. Если для этого случая существует специальный стандарт, то никогда не следует ограничиваться только настоящим стандартом.
C.3 Замечания по специальным разделам и подразделам
5 МЭК 60601-1 является повсеместно принятым к использованию стандартом, охватывающим большинство вопросов безопасности, связанных с неимплантируемыми частями активных имплантируемых медицинских изделий.
7.1 Упаковка одноразового использования становится стерильной упаковкой после того, как сама упаковка и ее содержимое были стерилизованы. Требования стерильности приведены в 14.1.
7.2 Товарная упаковка обеспечивает защиту одноразовой упаковки при неблагоприятных условиях транспортирования и хранения, а также обеспечивает доступ к сопровождающей документации в месте применения. См. также 10.4.
8 В целом, информация, приводимая на маркировке активного имплантируемого медицинского изделия, должна быть полезна клиницисту при применении изделия для лечения. Дополнительная информация, не имеющая отношения к этой цели, может затруднить понимание информации, существенной для корректного использования изделия.
8.1 Требуемые предупреждения должны быть различимы с первого взгляда, не быть при этом слишком мелкими и не перекрываться другими предупреждающими надписями, чтобы избегать ненужного риска для пациентов.
8.2 При отказе ряда образцов имплантируемого изделия может стать очевидным, что должны быть предприняты действия по предотвращению нанесения вреда пациенту применением схожих изделий. Пострадавшие пациенты должны быть четко идентифицированы, чтобы избежать ненужных медицинских процедур. Это требует четкой идентификации как изделия, так и его компонентов без необходимости эксплантации изделия. Правильная идентификация компонентов зависит от того, насколько изготовитель поддерживает ведение систематических записей. Руководство по одному из подходов к ведению записей представлено в серии стандартов ИСО 9000 вместе со стандартами ИСО 13485 и ИСО 13488.
9 В общем, в маркировке на товарной упаковке следует строго избегать несущественной информации, которая снижает ясность важных сведений, требуемых настоящим стандартом.
9.1 Упаковка, содержащая источники радиации, может быть предметом дополнительного специального контроля.
9.3 Следует уделять внимание только той информации, которая необходима пользователю для идентификации изделия в процессе его выбора при проведении медицинской процедуры.
9.4 Может оказаться, что информации, требуемой на товарной упаковке в соответствии с другими подразделами стандарта, будет недостаточно для идентификации изделия и его характеристик.
9.5 Настоящее требование предназначено для обеспечения мер надлежащего обращения с изделием.
9.6 и 11.4 В принятом повсеместно к применению стандарте ИСО 5841-1 на имплантируемые электрокардиостимуляторы уже установлены требования к формату даты. ИСО 8601 устанавливает международно принятый формат даты.
9.8 Для определения точного содержимого не обязательно вскрывать упаковку. После вскрытия в упаковке должны быть в наличии все специальные принадлежности, необходимые пользователю для применения изделия. Это позволит защитить разупакованное, но не использованное изделие от повреждения.
9.9 После вскрытия упаковки вложенное изделие должно использоваться с принадлежностями, поставляемыми отдельно, которые врач намерен использовать в ходе имплантации. Это позволит также сохранить распакованное, но не использованное изделие в пригодном виде до тех пор, пока не будут найдены подходящие принадлежности.
9.11 См. раздел 25 и 26.2 на предмет обсуждения исключительных условий транспортирования и хранения.
9.12 и 11.10 Какое-либо указание на специальное назначение изделия помогает избежать применения изделия пациенту, для которого данное изделие не предназначено.
10.1 Упаковка должна гарантировать то, что изделие не подвергалось воздействию опасных условий во время транспортирования и хранения. Допустимые пределы зависят от дизайна конструкции рассматриваемого изделия.
10.2 Атмосферная влажность во время транспортирования не должна вызывать ухудшения нанесенной информации, предназначенной пользователю. Это требование основано на соответствии подразделу 4.10 МЭК 60601-1 и на испытании Cb по МЭК 60068-2-56.
10.3 Испытание на влажное истирание определяет требование постоянства неуничтожимости и различимости маркировки. Это требование основано на требовании соответствия подразделу 6.1 МЭК 60601-1.
10.4 Фундаментальным принципом является то, что изделие соответствует функции, указанной изготовителем и заявленной пользователю в маркировке и сопровождающей документации. Это требование считается нарушенным, если информация неправильно соотносится с изделием.
11 В целом маркировка на стерильной упаковке должна быть ограниченной по содержанию, чтобы избежать несущественной информации, которая снижает ясность представления информации, требуемой настоящим стандартом.
11.7 Важно, чтобы пользователь мог проверить, все ли у него в наличии из того, что требуется, непосредственно перед имплантацией изделия без необходимости предварительного вскрытия стерильной упаковки. Если упаковка остается открытой в течение неопределенного периода времени перед имплантацией, то это может вызвать загрязнение изделия или его повреждение.
11.8 Это требование позволяет подтвердить окончательно виды соединений перед вскрытием упаковки. (Например, на тот случай, если стерильная упаковка отделена от сопровождающей документации). Если же упаковка остаётся открытой в течение неопределенного периода времени перед имплантацией, то это может вызвать загрязнение изделия или его повреждение.
12.1 ИСО 11607 является общим стандартом, уточняющим требования к материалам одноразового применения и контейнерам многоразового использования для хранения упаковки изделий для окончательной стерилизации.
13.1 Такая маркировка обеспечивает идентификацию изделия при его извлечении (эксплантации). Некоторые имплантируемые части могут быть слишком мелкими, чтобы на них могла быть нанесена маркировка. Некоторые принадлежности (например, в виде прилагаемых инструментов) могут не нуждаться в нанесении номера партии или серийного номера. Это требование основано на соответствии требованию МЭК 60601-1, подраздел 6.1.
13.2 Это требование позволяет пользователю группировать элементы при анализе данных по сроку их долговечности. Характеристики батареи питания, которые изначально номинально равноценны, часто показывают существенные различия по долговечности к концу срока службы имплантата.
13.3 Настоящий подраздел затрагивает фундаментальный принцип: любое используемое изделие может быть идентифицировано без проведения хирургической операции и без необходимости в специализированном оборудовании, характерном для компании-изготовителя или модели изделия. На практике может оказаться неудобным маркировать мелкие неактивные имплантируемые изделия. Сегодня высоким уровнем развития технологий считается идентификация изготовителя и модели изделия с помощью рентген-контрастных символов-меток, если изделие содержит источник энергии. Считывание рентген-контрастного символа позволяет выбрать подходящее телеметрическое устройство, которое, в свою очередь, может позволить идентифицировать серийный номер имплантированного изделия.
13.4 Безопасное применение каждого изделия требует (с учетом соответствующих допущений об уровне обучения и знаний потенциального пользователя), чтобы к нему прилагалась некая ключевая информация. Насколько практично и допустимо, информация, требуемая для безопасного использования изделия, должна быть нанесена на само изделие. Если возможно, такая информация должна быть представлена в виде символов. При этом любые символы и их цветовая индикация должны соответствовать международным стандартам. Если соответствующего стандарта не существует, символы и цветовая индикация должны быть описаны в документации, предоставляемой изготовителем.
14.1 Во избежание ненужных перемещений и процессов предоперационной подготовки в клинике имплантируемые части активных имплантируемых медицинских изделий должны поставляться стерильными в одноразовой упаковке. Если другие части включены в одноразовую упаковку, они также должны быть стерильными во избежание загрязнения имплантируемых частей. Материалу, содержащемуся в герметично запечатанном контейнере в течение всего срока службы изделия, не требуется быть стерильным.
14.2 Поскольку особым требованием к имплантату является то, чтобы он не заносил инфекцию в организм больного, то не должно быть излишнего введения веществ со свободными твердыми примесями ("стерильная грязь"). Метод оценки основан на сопоставлении результатов испытаний с установленными количественными пределами содержания примесей. Подходит любое средство измерения, использующее данный метод. Испытания, основанные на стандартных испытаниях на наличие частиц, описаны в Британской фармакопее.
14.3 В контексте настоящего стандарта термин "биосовместимость" означает высокую степень уверенности в том, что имплантируемый материал будет функционировать без причинения вреда организму или не повреждаясь, в свою очередь, со стороны организма. Биологические испытания, покрывающие срок службы имплантированного изделия, невыполнимы. Соответственно подтверждение биосовместимости можно получить только на основании обобщения существующих опытных данных и/или признанных процедур биологической оценки (например, путем применения какой-либо из частей ИСО 10993).
16.2 Наличие непрерывных малых прямых токов из имплантируемых электродов может вызвать повреждение тканей или коррозию электрода. Безопасным значением может быть ток, пониженный до 0,1 мкА для малых электродов. Метод испытаний должен быть применим даже к изделиям, используемым для контроля физиологических процессов.
16.3 Электрическая изоляция должна быть адекватной, даже если изоляция проницаема для жидких сред организма. Существует функциональная взаимосвязь между временем нарастания прикладываемого тестового напряжения, напряжением и числом повторений испытания, но эта взаимосвязь не может быть определена для всего множества изделий, охватываемых настоящим стандартом. Отобранные значения предназначены для того, чтобы установить осуществимое испытание, дающее значительные результаты. Другие части ИСО 14708 могут устанавливать другие виды испытаний на оценку.
17 Из-за хорошего теплового контакта между активным имплантируемым медицинским изделием и тканями пациента повышение температуры на 2 °С на внешней поверхности имплантируемого изделия требует значительного рассеяния энергии с обычного имплантируемого источника энергии. Могут требоваться более сложные испытательные методики и механизмы защиты, если имплантированный источник энергии может поставлять энергию с высокими значениями, как в случае имплантируемого дефибриллятора. Действительная температура поверхности изделия, работающего в особых условиях или в условиях единичного отказа, зависит от температуры тела пациента, которая является внешней для имплантата. Пределы температуры, установленные в настоящем стандарте, ниже соответствующих пределов, установленных МЭК 60601-1, ввиду повышенной трудности проведения корректирующих действий, если источник энергии имплантирован.
18.1-18.3 В настоящее время допустимо, что некоторые активные имплантируемые медицинские изделия могут содержать радиоактивные вещества. На момент издания настоящего стандарта неизвестно ни одного активного имплантируемого медицинского изделия, которое бы специально испускало радиоактивное излучение. К источникам радиоактивной энергии применимы только требования, описанные в настоящих разделах.
19.1 Большинство активных имплантируемых медицинских изделий разработаны для имплантации на срок в несколько лет. Таким образом, испытания, целью которых является определение времени до отказа изделия или получение информации о сроке службы изделия, часто невыполнимы. Приемлемой практикой является сокращение времени получения необходимых данных путем проведения ускоренных испытаний. На основании результатов такого испытания можно сделать заключение о параметрах срока службы при большинстве случаев благоприятных условий работы имплантируемого изделия и убедиться в том, что:
a) отказы, наблюдаемые при ускоренных испытаниях, схожи с таковыми, полученными при использовании в нормальных условиях;
b) допустима - с разумным уровнем достоверности - экстраполяция значений от ускоренных испытаний к нормальным условиям применения.
Опыт применения активных имплантируемых медицинских изделий за последние годы показывает, что уровень преждевременных отказов изделий достигает очень низкого значения, какого и ожидают пользователи. Таким образом, ускоренные испытания, используемые для оценки надежности активных имплантируемых медицинских изделий, не только должны соответствовать требованиям, описанным выше, но должны быть разработаны таким образом, чтобы достигать статистически значимого уровня типичных видов отказов менее 0,5% в год.
19.2 Важно, чтобы разряжение источника энергии активного имплантируемого медицинского изделия не вызывало прекращения его функционирования без предварительного предупреждения. Механизм предупреждения не должен выключаться из работы по вине различных видов терапевтических процедур, которые могут истощить источник энергии с разными скоростями.
19.3 Активное имплантируемое медицинское изделие должно оставаться безопасным при наступлении отказа. Из-за ограничений конструкции применяемых в настоящее время изделий считается, что анализ первого отказа может быть выполнен только разработчиком изделия. Методология анализа установлена настоящим стандартом. Требования к надежному программному обеспечению программируемых систем устанавливаются той же методологией.
19.4 Некоторые критерии традиционных фармакологических клинических исследований могут быть неприменимы к активным имплантируемым медицинским изделиям: например, распределения по возрастам и двойного слепого контроля. Область любого клинического исследования будет ограничена малой доступной целевой популяцией и относительно низким числом случаев целевой патологии.
20.2 Элементы цепи на рисунке 1 расположены таким образом, что энергия, подаваемая на изделие, когда последнее напрямую подключено к испытательному оборудованию через резистор 300 Ом, подобна энергии, подаваемой на изделие с помощью водителя ритма, когда пациент подвергается дефибрилляции с использованием внешних электродов дефибриллятора. При таком испытании следует избегать использования высокого напряжения, поставляемого непосредственно электродами дефибриллятора. Требования основаны на ИСО 5841-1, раздел 6 и рисунки 1 и 2.
Попытки дефибрилляции часто приходится повторять, и полярность подаваемого сигнала при этом не ограничена. Настоящий подраздел предназначен для определения такого практического уровня защиты, чтобы в большинстве случаев дефибрилляция не вызывала повреждений активного имплантируемого медицинского изделия. В целом невозможно обеспечить полную защиту активных имплантатов, содержащих полупроводники. Невидимые повреждения могут сократить срок службы полупроводниковых компонентов. Отсюда и соответствующие требования в 28.13.
21 Настоящий раздел предназначен для обеспечения разумного уровня защиты от идентифицируемых опасностей, таких как хирургическое вмешательство или назначение курса физиотерапии, в частности диатермии. Это требование может быть дополнено анализом случаев низкого уровня защиты, приведенным в разделе 27. В целом невозможно обеспечить полную защиту активных имплантатов, содержащих полупроводники. Невидимые повреждения могут сократить срок службы полупроводниковых компонентов. Отсюда и соответствующие требования в 28.13.
22 Следует обратить внимание, что данное требование адресовано к воздействию только диагностического ультразвука. В настоящем стандарте воздействие ультразвука на активное имплантируемое медицинское изделие в терапевтических дозах определяется требованием о предупреждающих надписях (см. 28.14).
23.1 Известно, что данное требование является более строгим, чем аналогичное требование МЭК 60601-1. Программаторы, переносимые вручную, и портативные анализаторы могут подвергнуться тяжелым механическим ударам во время переноски кем-либо иным, кроме опытного пользователя. При таких ударных воздействиях полученное повреждение не сразу проявится для пользователя, но поврежденное изделие может неверно настроить имплантат или дать ложный анализ имплантированного изделия, что впоследствии приведет к ненужному извлечению имплантата.
23.2 Настоящий подраздел устанавливает минимальный стандарт прочности активных имплантируемых медицинских изделий. Руководство, представленное в МЭК 60068, подразумевает, что данное испытание на воздействие случайной вибрации является более уместным, чем испытание на синусоидальную вибрацию, описанное в другой части названного стандарта, которое было ранее определено для оценки имплантируемых кардиостимуляторов.
Диапазон частот определяется с учетом использования изделия. Нижний предел частоты расширен до 5 Гц в связи с тем, что имплантированное изделие может подвергаться воздействию вибрации низкой частоты, которая может вызывать смещение внутренних компонентов относительно друг друга. Верхний предел частоты ограничен 150 Гц, потому что тело пациента наделено способностью к защите изделия от воздействий вибрации высокой частоты, которые в противном случае были бы существенны для малых электронных изделий.
Защита изделия во время транспортирования и хранения обеспечивается соответствующей конструкцией упаковки.
23.3 Известно, что имплантированные проводники и катетеры иногда подвергаются воздействию растягивающих сил после имплантации. Эти силы, возможно, вызваны движениями тела во время занятий спортом или физическими силами, прикладываемыми непосредственно на тело, например, при несчастных случаях.
23.4 и 23.5 Эти требования предназначены для обеспечения того, чтобы было проведено адекватное исследование для обеспечения защиты от усталостных отказов имплантированных электродов и катетеров.
23.6 Настоящий стандарт оставляет метод обеспечения безопасности соединения на усмотрение изготовителя. Таким образом, от изготовителя требуется определить совместимые части соединений (см. 9.9 и 28.9) так, чтобы определенные части были выбраны для испытания на подтверждение того, что имплантируемые части соединения надежно и прочно соединены на случай воздействия сил растяжения.
В требовании настоящего стандарта по оценке соответствия дополнительно к силе растяжения прикладывается крутящий момент, поскольку имплантированные части соединения могут подвергнуться воздействию кручения ввиду медленного смещения имплантированного изделия в течение срока службы.
В подразделе приведены рекомендуемые значения крутящего момента и сосредоточенных сил для оценки прочности соединения, однако разработка испытательного оборудования и средств мониторинга испытания остаются за изготовителем изделия или независимой испытательной лабораторией.
24 Эти требования применяются только к неимплантируемым частям, которые могут подвергаться воздействию условий окружающей среды лечебного учреждения. Предполагается, что имплантированные части достаточно защищены от воздействия электростатических потенциалов их упаковкой и тем, что с ними работают только в контролируемых условиях.
25 Нижний предел границы соответствует высоте приблизительно 3000 м (т.е. на 30 кПа ниже нормального атмосферного давления). Верхний предел соответствует глубине приблизительно 5 м под водой (т.е. на 50 кПа выше нормального атмосферного давления).
26.2 Известно, что некоторые имплантаты не выдерживают замораживания. Это следует учитывать в соответствующих специальных стандартах (соответствующая часть ИСО 14708). Любое изделие, которое не может быть подвергнуто воздействию широкого диапазона температур, указанных в данном подразделе, требует размещения дополнительных предупреждающих надписей на упаковке (см. 9.11). Если повреждения, вызванные замораживанием, проявляются не сразу, возможно потребуются специальные индикаторы температуры на упаковке.
27 Данная общая процедура предназначена для обеспечения защиты от электромагнитных воздействий. В последующих частях ИСО 14708 это требование, вероятно, будет дополнено детальными испытаниями, применимыми для активных имплантируемых медицинских изделий специальных типов.
28.6 Код, указанный в 13.3, следует разъяснить пользователю.
28.18 Имплантируемое изделие можно использовать повторно при условии, что "восстановление" отвечает всем требованиям настоящего стандарта. Данное условие касается фундаментального принципа в отношении как информации, приводимой на этикетке упаковки, так и в сопровождающей документации.
28.19 На определение срока службы изделия не должны влиять ни настройки имплантата пользователем в рамках практического клинического применения, ни физиология пациента, ни физическое поведение пациента.
28.23 Некоторые источники, которые могут представлять опасность для электронных имплантатов различных типов, обычно имеют предупреждающие надписи, предназначенные только "пациентам с электрокардиостимуляторами". Эти предупреждения могут, поэтому, относиться к другим активным имплантируемым медицинским изделиям, хотя без специальной консультации пациент может игнорировать предупреждения, если у него нет электрокардиостимулятора.
Поэтому данный подраздел не предназначен для того, чтобы пациент пренебрегал надписями; требования подраздела предназначены для того, чтобы пациент с активным имплантируемым медицинским изделием, отличным от электрокардиостимулятора, осознавал, что предупреждение, адресованное пациентам с электрокардиостимуляторами, может быть важным и для него самого.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
ИСО 8601:1998 | - | * |
ИСО 11607:1997 | IDT | ГОСТ Р ИСО 11607-2003 "Упаковка для медицинских изделий, подлежащих финишной стерилизации. Общие требования" |
ИСО 14155:1996 | IDT | ГОСТ Р ИСО 14155-1-2008 "Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования" |
ИСО 15223:2000 | IDT | ГОСТ Р ИСО 15223-2002 "Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации" |
МЭК 60068-2-14:1996 | - | * |
МЭК 60068-2-32:1990 | - | * |
МЭК 60068-2-47:1999 | - | * |
МЭК 60068-2-64:1993 | - | * |
МЭК 60601-1:1988 | MOD | ГОСТ Р 50267.0-92 "Изделия медицинские электрические. Часть 1. Общие требования безопасности" |
МЭК 60601-1-1:1992 | - | * |
МЭК 60601-1-2:1993 | - | * |
МЭК 60601-1-4:1996 | - | * |
МЭК 60601-2-27:1994 | MOD | ГОСТ Р 50267.27-95 "Изделия медицинские электрические. Часть 2. Частные требования безопасности к электрокардиографическим мониторам" |
МЭК 61000-4-2:1995 | - | * |
* Соответствующий национальный стандарт отсутствует. До его утверждения рекомендуется использовать перевод на русский язык данного международного стандарта. Перевод данного международного стандарта находится в Федеральном информационном фонде технических регламентов и стандартов. Примечание - В настоящей таблице использованы следующие условные обозначения степени соответствия стандартов: - MOD - модифицированные стандарты; - IDT - идентичные стандарты. | ||
________________
| ||
Библиография
[1] ISO 5841-1:1989, Cardiac pacemakers - Part 1: Implantable pacemakers
[2] ISO 11134:1994, Sterilization of health care products - Requirements for validation and routine control - Industrial moist heat sterilization
[3] ISO 11135:1994, Medical devices - Validation and routine control of ethylene oxide sterilization
[4] ISO 11137:1995, Sterilization of health care products - Requirements for validation and routine control - Radiation sterilization
[5] ISO 9001: -, Quality systems - Requirements
________________
Пересмотр ISO 9001:1994, ISO 9002:1994 и ISO 9003:1994.
[6] ISO 9002:1994, Quality systems - Model for quality assurance in production, installation and servicing
[7] ISO 10993-1:1997, Biological evaluation of medical devices - Part 1: Evaluation and testing
[8] ISO 13485:1996, Quality systems - Medical devices - Particular requirements for the application of ISO 9001
[9] ISO 13488:1996, Quality systems - Medical devices - Particular requirements for the application of ISO 9002
[10] ISO/TR 14283:1995, Implants for surgery - Fundamental principles
[11] IEC 60068-2-17:1994, Environmental testing - Part 2-17: Tests - Test Q: Sealing
[12] IEC 60068-2-33:1978, Environmental testing - Part 2-33: Tests. Guidance on change of temperature tests
[13] IEC 60068-2-56:1988, Environmental testing - Part 2-56: Tests. Test Cb: Damp heat, steady state, primarily for equipment
[14] British Pharmacopeia, Vol. 2 Appendix XII: A163 Limit test for particular matter. London, HMOS, 1993, ISBN 0 11 321543 6
_____________________________________________________________________
УДК 616.126.3-089.28:006.354 OКC 11.040.40 P26
Ключевые слова: имплантируемое медицинское изделие, имплантаты хирургические, безопасность, маркировка
_____________________________________________________________________
Электронный текст документа
и сверен по:
, 2014
