
ГОСТ ISO/TR 17276-2016
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ПРОДУКЦИЯ ПАРФЮМЕРНО-КОСМЕТИЧЕСКАЯ
Аналитический подход для методов скрининга и количественного определения тяжелых металлов в косметике
Perfume and cosmetic products
Analytical approach for screening and quantification methods for heavy metals in cosmetics
МКС 71.100.70
Дата введения 2025-01-01
с правом досрочного применения
Предисловие
Цели, основные принципы и общие правила проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0 "Межгосударственная система стандартизации. Основные положения" и ГОСТ 1.2 "Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены"
Сведения о стандарте
1 ПОДГОТОВЛЕН республиканским унитарным предприятием "Белорусский государственный институт метрологии" (БелГИМ) на основе собственного перевода на русский язык англоязычной версии документа, указанного в пункте 5
2 ВНЕСЕН Госстандартом Республики Беларусь
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации по результатам голосования в АИС МГС (протоколом от 20 апреля 2016 г. N 87-П)
За принятие проголосовали:
Краткое наименование страны по МК (ИСО 3166) 004-97 | Код страны по МК (ИСО 3166) 004-97 | Сокращенное наименование национального органа по стандартизации
|
Армения | AM | ЗАО "Национальный орган по стандартизации и метрологии" Республики Армения |
Беларусь | BY | Госстандарт Республики Беларусь |
Казахстан | KZ | Госстандарт Республики Казахстан |
Киргизия | KG | Кыргызстандарт |
Россия | RU | Росстандарт |
Таджикистан | TJ | Таджикстандарт |
Туркмения | TM | Главгосслужба "Туркменстандартлары" |
Узбекистан | UZ | Узстандарт |
(Поправка. ИУС № 7-2024).
4 Приказом Федерального агентства по техническому регулированию и метрологии от 27 декабря 2023 г. N 1685-ст межгосударственный стандарт ГОСТ ISO/TR 17276-2016 введен в действие в качестве национального стандарта Российской Федерации с 1 января 2025 г. с правом досрочного применения
5 Настоящий стандарт идентичен международному документу ISO/TR 17276:2014* "Косметика. Аналитический подход для методов скрининга и количественного определения тяжелых металлов в косметике" (Cosmetics - Analytical approach for screening and quantification methods for heavy metals in cosmetics, IDT).
Международный документ разработан техническим комитетом ISO/TC 217 "Косметика" Международной организации по стандартизации (ISO).
Наименование настоящего стандарта изменено относительно наименования указанного международного документа для приведения в соответствие с ГОСТ 1.5-2001 (подраздел 3.6)
6 ВВЕДЕН ВПЕРВЫЕ
Информация о введении в действие (прекращении действия) настоящего стандарта и изменений к нему на территории указанных выше государств публикуется в указателях национальных стандартов, издаваемых в этих государствах, а также в сети Интернет на сайтах соответствующих национальных органов по стандартизации.
В случае пересмотра, изменения или отмены настоящего стандарта соответствующая информация будет опубликована на официальном интернет-сайте Межгосударственного совета по стандартизации, метрологии и сертификации в каталоге "Межгосударственные стандарты"
ВНЕСЕНА поправка, опубликованная в ИУС № 7, 2024 год
1 Область применения
Настоящий стандарт устанавливает наиболее распространенные типовые подходы к выполнению скрининга и количественного определения важнейших тяжелых металлов на этапе подготовки сырья, а также после получения готовой продукции. Он охватывает широкий круг методов, начиная с традиционных, основанных на использовании колориметрических реакций, которые могут быть реализованы без применения дорогостоящего оборудования, и заканчивая наиболее высокотехнологичными, такими как масс-спектрометрия с индуктивно связанной плазмой (ИСП-МС), позволяющая обнаруживать вещества, уровень содержания которых выражается в микрограммах на килограмм. В настоящем стандарте рассмотрены преимущества и недостатки каждого из этих аналитических методов, что позволяет пользователю выбрать для себя наиболее приемлемый подход.
Настоящий стандарт не предназначен для того, чтобы установить или рекомендовать значения предельно допустимой концентрации тяжелых металлов в сырье или готовой продукции. Эти значения должны устанавливаться соответствующими регулирующими органами.
Примечание 1 - Широко используемый термин "тяжелые металлы" не имеет единого определения.
Примечание 2 - Элементы, рассматриваемые как тяжелые металлы в законодательстве одной страны, могут не рассматриваться как таковые в законодательстве другой страны.
2 Принципы исследования
2.1 Планирование
Прежде всего применяемые подходы разделяют на скрининг и количественное определение общего содержания тяжелых металлов. Выполнение анализа тяжелых металлов требует не только обладания техническими знаниями и навыками, но и зачастую наличия дорогостоящего оборудования и строгого соблюдения условий подготовки проб, в особенности если тяжелые металлы подлежат количественному определению. Скрининг помогает принять решение о том, требуют ли уровни содержания тяжелых металлов дальнейшего определения с применением более точных количественных методов.
Анализ содержания тяжелых металлов в косметической продукции и косметическом сырье предполагает выбор подходящего метода подготовки проб и метода обнаружения. Условия аналитических испытаний определяются необходимым сочетанием метода подготовки проб и метода обнаружения с соответствующими данными валидации.
К методам подготовки проб относятся:
- выщелачивание;
- минерализация.
К методам и способам обнаружения относятся:
- колориметрическая реакция; [3]-[8];
- рентгеновская флуоресценция (РФ);
- атомно-абсорбционная спектрометрия (AAС);
- оптико-эмиссионная спектроскопия с индуктивно связанной плазмой (ИСП-ОЭС), известная как атомно-эмиссионная спектроскопия с индуктивно связанной плазмой (ИСП-АЭС);
- масс-спектрометрия с индуктивно связанной плазмой (ИСП-МС).
Эти методы, как правило, не учитывают различия между органическими и неорганическими соединениями, а также соединениями того или иного исследуемого элемента. Например, они не дифференцируют металлическую ртуть и фенилртутные соединения. Кроме того, они оставляют без внимания возможные неодинаковые валентные состояния элемента, как в случае с хромом (III) и хромом (VI). Если сведения такого рода представляют особый интерес, для их уточнения следует воспользоваться соответствующими средствами, например прибегнуть к помощи ИСП-МС в сочетании с хроматографией.
Типовой подход к выполнению скрининга и количественного определения тяжелых металлов в сырье и готовой продукции представлен в приложениях A, B и C. Подходы, отличные от описанного в указанных приложениях, также могут оказаться эффективными.
2.2 Выбор исследуемого вещества
Скрининг и количественное определение тяжелых металлов могут выполняться как в сырье, так в готовой продукции.
Поскольку тяжелые металлы свободно встречаются в природе, некоторые виды сырья, в частности некоторые неорганические материалы, могут содержать примеси тяжелых металлов естественного происхождения. Знание источника и характеристик сырьевых материалов обеспечивает действенный подход к установлению контроля над уровнями содержания тяжелых металлов в готовой продукции. Осуществление мониторинга на этапе сырья позволяет избежать использования сильно загрязненного сырья и является эффективным средством контроля содержания тяжелых металлов в готовой продукции.
2.3 Подготовка пробы
2.3.1 Общие положения
Многие методы элементного анализа предусматривают перевод пробы в жидкую форму. Порядок приготовления пробы зависит от матрицы косметического сырья или продукции. Методы приготовления проб обычно подразделяются на два вида: выщелачивание и минерализация.
2.3.2 Выщелачивание
Выщелачивание - это метод подготовки проб, предназначенный для определения количества тяжелых металлов, экстрагируемых из пробы в условиях кислой среды. Принцип выщелачивания заключается в имитации воздействия пищеварительных жидкостей или пота, направленных на высвобождение тяжелых металлов, возможно присутствующих в исследуемой продукции. Это позволяет оценить количество тяжелых металлов, которое фактически попадает в организм человека.
2.3.3 Метод минерализации
Метод минерализации - это метод подготовки проб, предназначенный для определения общего количества тяжелых металлов, содержащихся в пробе. Полная минерализация позволяет оценить количество тяжелых металлов, воздействующих на организм человека в наихудшем возможном случае. Кроме того, выполнение полной минерализации матрицы сокращает помехи при обнаружении, особенно в случае использования ИСП-МС.
В некоторых случаях пробы просто нагревают до получения золы (сухое озоление), чтобы избавиться от органических веществ. Сухое озоление может выполняться с применением нитрата магния в качестве озоляющей добавки [9], [10]. Могут использоваться и другие виды озоляющих добавок, например смесь сульфата магния и серной кислоты [8]. Ввиду сложности матрицы косметической продукции после озоления нередко остаются нерастворимые вещества и требуется проведение дополнительных этапов минерализации.
Минерализация проб осуществляется путем нагревания, обычно с добавлением кислоты одного вида, иногда с добавлением нескольких кислот (влажная минерализация), очень редко с использованием щелочей (сплавление) в открытых или закрытых емкостях таким образом, чтобы добиться их полного или почти полного растворения. Это зачастую требует строгого соблюдения установленных требований и мер предосторожности с учетом вероятной возгонки отдельных металлов (таких, как кадмий, мышьяк или ртуть), чтобы обеспечить надлежащую степень извлечения [8], [11].
В соответствии с последними тенденциями применяется минерализация в закрытых емкостях с применением микроволнового излучения, что позволяет сократить потери летучих элементов, а также повысить эффективность регулярно выполняемых анализов. Правильный подбор используемых кислот - это существенный фактор, который следует учитывать при выполнении полной минерализации пробы. Считается, что при исследованиях косметической продукции для минерализации соединений кремния чрезвычайно результативно использование плавиковой кислоты (HF). Применение плавиковой кислоты требует последующей обработки пробы борной кислотой для маскирования остатков HF. Помимо этого, для минерализации проб могут применяться азотная, соляная, серная и другие кислоты. Каждая из кислот, включая HF, имеет свои преимущества, поэтому зачастую они используются совместно для достижения наиболее полной минерализации. Существует большое количество публикаций, посвященных анализу тяжелых металлов, включая оценку соответствующих методов минерализации. Так, известен метод минерализации, для которого в недавнем времени были опубликованы результаты межлабораторных сличительных испытаний для свинца, кадмия и ртути в различных видах готовой продукции, содержащей неорганические материалы. Упомянутый метод описывает процесс минерализации с использованием азотной кислоты в смеси с соляной кислотой в закрытом сосуде при повышенном давлении и с нагреванием приблизительно до 200°C. Он подробно определяет условия проведения анализа, соблюдение которых необходимо для получения воспроизводимых результатов [12]. В другом исследовании, предпринятом официальным органом, было проведено сравнение сходных аналитических результатов, полученных с использованием азотной кислоты и азотной кислоты в смеси с HF. На некоторых видах косметической продукции минерализация с помощью азотной кислоты показала более низкие результаты, чем с помощью смеси азотной кислоты и HF [13]. Несмотря на это, рекомендуется по возможности избегать применения плавиковой кислоты как из гигиенических соображений, так и из соображений безопасности.
2.4 Методы и способы обнаружения
2.4.1 Колориметрическая реакция
Соответствующая методика рассматривает колориметрический анализ как испытания на обнаружение тяжелых металлов, образующих нерастворимые сульфиды с окраской от желтой до темно-коричневой в среде с показателем pH от 3,0 до 3,5. Обычно объектом таких испытаний является сырье. К элементам, поддающимся обнаружению по этому методу, относятся например свинец, висмут, медь, кадмий, сурьма, олово и ртуть [8]. Нерастворимые сульфиды, получаемые в процессе реакции в разбавленных растворах, дают темную окраску в силу их коллоидной дисперсии. В качестве источника сульфидных ионов, как правило, выступает либо сульфид натрия, либо тиоацетамид. Насыщенность окраски возрастает пропорционально концентрации тяжелых металлов. Количество тяжелых металлов выражается как концентрация свинца в сравнении с концентрацией в стандартном растворе свинца. Преимущество подобной методики заключается в том, что она может быть реализована без применения дорогостоящего оборудования. Колориметрические испытания применимы только к неокрашенным растворам проб, не содержащим нерастворимых включений. Оценка количества извлеченных элементов должна осуществляться точными, соответствующими способами, в особенности если для получения соответствующих растворов применялось сухое озоление. Использование данной методики не позволяет обнаруживать присутствие селена и хрома. Кроме того, в присутствии цинка может образовываться белый осадок, также затрудняющий получение правильных результатов. По этой причине важно подтвердить надежность результатов испытаний при помощи соответствующей процедуры валидации.
Если между раствором пробы и стандартным раствором наблюдается существенное расхождение в получаемых оттенках цветов, то в этом случае целесообразно испробовать иные методы обнаружения.
Примечание - Способы проведения колориметрических испытаний описаны в ряде нормативных сборников по косметике и фармацевтике [3]-[7], таких как японские стандарты по лекарственно-подобным веществам (JSQI) [3] и Европейская фармакопея [4]. Кроме того, некоторую полезную практическую информацию и в особенности ту, что касается описаний на английском языке, можно найти в японских стандартах по косметическим ингредиентам [5] и Японском кодексе по косметическим ингредиентам [6], несмотря на то, что формально они уже утратили силу и большая часть их положений была обобщена в рамках JSQI.
2.4.2 Рентгеновская флуоресценция
2.4.2.1 Общие положения
При облучении пробы рентгеновским излучением с энергией, превышающей определенный предел, внутренние электроны в атомах приходят в возбуждение и покидают свои места, что приводит к появлению дырок. Соответственно, в образовавшиеся дырки затем попадают периферийные электроны, а избыточная энергия, соответствующая разности энергетических уровней, высвобождается в виде электромагнитных волн рентгеновского диапазона, обозначаемых как "рентгеновская флуоресценция". Поскольку разность энергетических уровней для каждого элемента уникальна, наблюдаемую рентгеновскую флуоресценцию еще называют "характеристическим рентгеновским излучением". Идентификация элемента становится возможной исходя из его рентгеновского спектра, а его концентрация в исследуемой пробе может быть определена на основе интенсивности рентгеновского излучения. Преимуществом данного способа является его неразрушающий характер. Измерения в этом случае могут выполняться на пробах, представленных в самой разнообразной форме: как твердые тела, как жидкость или как порошок. Такие измерения отличает легкость и быстрота выполнения, без сложного процесса подготовки проб. Со сложностями приходится столкнуться на этапе количественного или полуколичественного анализа, поскольку данный метод сильно подвержен влиянию матрицы, а, следовательно, для получения достоверных результатов требуется валидация метода или внесение в результаты соответствующих поправок. Для некоторых элементов обеспечить достаточную чувствительность рентгенофлуоресцентного метода не представляется возможным, особенно если используется портативное оборудование.
2.4.2.2 Типы оборудования
Применяемое оборудование можно грубо разделить на два типа в соответствии с реализованными в них принципами детектирования: с дисперсией по энергии и с дисперсией по длинам волн. Каждый из типов имеет свои особенности, а, следовательно, при выборе того или иного типа необходимо учитывать их преимущества и недостатки.
2.4.2.2.1 Энергодисперсионный тип
Представляет собой полупроводниковый детектор. Поскольку детектор сам по себе обладает неким энергетическим разрешением, конфигурация оборудования может быть упрощена по сравнению с волнодисперсионным типом. Как следствие, энергодисперсионное оборудование представлено более компактными приборами, чем аналогичное волнодисперсионное. Недостатками по сравнению с ним являются малая чувствительность и низкая разрешающая способность. Элементы, поддающиеся обнаружению, обычно варьируются от натрия до урана, с тенденцией повышения чувствительности для более легких элементов.
2.4.2.2.2 Волнодисперсионный тип
Преимуществами подобного оборудования являются высокие чувствительность и энергетическое разрешение, недостатком - значительные габариты. Основные элементы, поддающиеся обнаружению, варьируются от бериллия до урана. Флуоресцентное рентгеновское излучение, испускаемое пробой, проходит сквозь щелевую диафрагму для формирования параллельного светового потока. Затем оно попадает на аналитическую кристаллическую призму для дифракции, чтобы обеспечить поступление на детектор определенной длины волны.
Существует несколько типов аналитических призм. Кристаллическая призма с соответствующими интервалами между гранями выбирается в зависимости от анализируемого диапазона длины волны. Что касается рентгеновского детектора, то, как правило, для более легких элементов (от бериллия до скандия) используется пропорциональная счетная трубка, а для обнаружения флуоресцентного рентгеновского излучения элементов с длиной волны в диапазоне от 0,2 до 0,3 нм или менее (от титана до урана) служит сцинтилляционный счетчик.
2.4.3 Атомно-абсорбционная спектрометрия (ААС) [15]
2.4.3.1 Общие положения
Электроны атомов в атомизаторе могут быть приведены в возбужденное состояние за время, не превышающее нескольких наносекунд, за счет поглощения известного количества энергии излучения с заданной длиной волны. Количество энергии, требуемое для конкретного электронного перехода, у всех элементов различается. Как правило, каждое значение длины волны соответствует только одному элементу. Сигнал в отсутствие пробы и с пробой в атомизаторе измеряется детектором, и отношение между двумя значениями (абсорбция) преобразуется в значение концентрации аналита согласно закону Бугера - Ламберта - Бера.
Метод требует наличия растворов стандартных образцов с известным содержанием аналита для определения отношения между измеренной абсорбцией и концентрацией аналита, что связывает его с законом Бугера - Ламберта - Бера.
Оборудование для ААС включает в себя источник излучения, атомизационную камеру, монохроматор, детектор и считывающее устройство. Для анализа атомарных составляющих пробы она нуждается в атомизации. К наиболее распространенным видам атомизаторов на сегодняшний день относятся пламенные атомизаторы и атомизаторы с графитовой кюветой.
ААС является чрезвычайно широко используемым методом, обеспечивающим хорошую чувствительность и высокую специфичность. Определение некоторых элементов в присутствии азотной кислоты при высоком содержании железа, алюминия и кремния может затрудняться помехами. К основным недостаткам ААС относятся необходимость полного растворения пробы, обусловленная моноэлементным характером данного метода (исключение составляет особый случай применения ААС с использованием графитовой печи и вводом проб в твердой фазе), а также относительно высокая стоимость исследования.
2.4.3.2 Пламенная ААС
При использовании пламенной ААС раствор пробы вводится в пламя ацетилена и газа-окислителя, в качестве которого могут выступать воздух или оксид азота, что позволяет атомизировать исследуемые элементы. Пламенная ААС обеспечивает высокую чувствительность при обнаружении щелочных и щелочноземельных металлов.
2.4.3.3 ААС с генерацией гидридов (ГГ-ААС)
2.4.3.4 ААС с использованием графитовой печи (ГП-ААС)
При использовании ААС с графитовой печью жидкая или твердая проба вводится в графитовую трубку, где выпаривается за счет электрического подогрева, а остатки озоляются. Для обеспечения надлежащей повторяемости, точности и высокой чувствительности рекомендуется использовать графитовую трубку с платформой. Если целевые элементы обладают повышенной летучестью, то для уменьшения потерь этих элементов при нагревании применяются модификаторы матрицы. При дальнейшем нагревании до очень высоких значений температур, элементы, присутствующие в остатках подвергаются атомизации. На этом этапе может быть измерено ослабление излучения лампы в узком объеме графитовой кюветы. ГП-ААС обычно обеспечивает более высокую чувствительность по сравнению с пламенной ААС по отношению к большому числу элементов, что требует внесения поправки на фон ввиду использования высоких температур. В качестве такой фоновой коррекции часто применяется зеемановская или дейтериевая коррекция.
2.4.3.5 ААС по методу "холодного пара" (ХП-ААС)
Анализ ртути ввиду ее специфических физико-химических свойств может предполагать использование специально разработанных методик подготовки проб и требует особого порядка приготовления пробы либо применения особых методов исследования. Поскольку для атомизации ртути не следует поддерживать высокие значения температур, это ее качество нередко используется при выполнении исследований с применением так называемого метода "холодного пара". Атомизация ртути происходит либо путем ее восстановления соответствующими восстанавливающими агентами, такими как дихлорид олова, либо путем нагревания. Вслед за нагреванием ртути иногда выполняется амальгамация ею золота, что позволяет избирательно вводить ртуть в ячейку атомизации с целью повышения чувствительности. На рынке представлен целый ряд приборов для анализа ртути, использующих описанное свойство этого элемента [10], [16], [17].
2.4.4 Индуктивно связанная плазма (ИСП)
2.4.4.1 Общие положения
ИСП предоставляет превосходные возможности возбуждения или ионизации элементов в силу высоких значений температур, достигаемых плазмой [18]-[20]. При включении горелки ИСП создается интенсивное электромагнитное поле. В горелке воспламеняется нагнетаемый газообразный аргон и образуется плазма. Поток аргона, необходимый для поддержания плазмы, имеет высокие значения расхода (~20 л/мин), а значения температуры достигают 7000 K или более. В большинстве случаев раствор пробы при помощи перистальтического насоса вводится в распылитель для получения аэрозоля. Аэрозоль поступает непосредственно в плазму аргоновой горелки, сразу же сталкиваясь с электронами и заряженными ионами плазмы так, что элементы, содержащиеся в пробе, превращаются в ионы. Молекулы пробы распадаются на соответствующие атомы, которые теряют электроны, что вызывает световое излучение с характеристическими длинами волны присутствующих в пробе элементов. Используемые детекторы бывают типов МС или ОЭС. Детекторы МС обнаруживают ионизированные атомы на основе отношения масса/заряд; детекторы ОЭС реагируют на свет, излучаемый возбужденными атомами [21].
2.4.4.2 ИСП-ОЭС
Если установка с ИСП оборудована оптическим спектрометром (ИСП-ОЭС), то концентрация элемента в исследуемой пробе определяется по интенсивности такого излучения. Метод ИСП-ОЭС отличается высокой чувствительностью, однако при его использовании необходимо учитывать явление спектральной интерференции (наличие нескольких линий испускания, как в случае с железом). Применение нескольких поправочных уравнений позволяет ослабить влияние данного явления.
2.4.4.3 ИСП-МС
Если установка с ИСП оборудована масс-детектором (ИСП-МС), то концентрация элемента в пробе определяется по распространенности соответствующих ионов (изотопов). Для ИСП-МС плазма должна иметь более высокую температуру, чтобы стимулировать образование ионов. Помехи при масс-детектировании могут быть полиатомными или изотопными (изобарными). В этом случае могут применяться поправочные уравнения, а в более современном оборудовании для снижения остроты или полного устранения данной проблемы используются коллизионные ячейки или анализаторы с высокой разрешающей способностью.
Значительным преимуществом ИСП-ОЭС и ИСП-МС являются многоэлементный характер данных методов и их линейный динамический диапазон. Основной недостаток - стоимость исследования, а также тот факт, что пробы, как правило, приходится вводить в виде раствора.
Приложение A
(справочное)
Колориметрическая реакция [3], [8]
A.1 Общие положения
К элементам, поддающимся такому обнаружению, относятся, например, свинец, висмут, медь, кадмий, сурьма, олово и ртуть.
Применяется следующий порядок испытаний.
1) Приготавливают испытуемый раствор (ИР) путем растворения пробы в воде, в которую добавлена уксусная кислота для получения значения pH в интервале от 3,0 до 3,5. В случае необходимости используют серную кислоту, азотную кислоту или иные кислоты, подходящие для разложения и удаления из пробы органических примесей. Тяжелые металлы в испытуемом растворе реагируют с добавляемым в него сульфидом натрия, образуя окрашенные нерастворимые сульфиды.
2) Аналогичные операции выполняют на стандартном растворе свинца.
3) Испытуемый раствор и стандартный раствор свинца по отдельности переносят в пробирки Несслера, а затем сравнивают окраску растворов путем наблюдения пробирок сверху и сбоку на белом фоне.
Использование титрованного раствора сульфида натрия в качестве колориметрического реактива описано в японских стандартах по лекарственноподобным веществам [3]. Сульфид натрия в растворе может быть заменен тиоацетамидом, в соответствии с требованиями Европейской фармакопеи 7.0, [4], [8], [7] и другими фармацевтическими нормами.
A.2 Растворы и стандартные растворы
A.2.1 Общие положения
В настоящем стандарте представлены только наиболее важные виды растворов. Все используемые реактивы должны иметь степень чистоты "чистый для анализа".
A.2.2 Стандартный раствор свинца (10 мг/л).
A.3 Оборудование
A.3.1 Пробирка Несслера, бесцветная цилиндрическая пробирка из тугоплавкого стекла с толщиной стенок от 1,0 до 1,5 мм и притертой пробкой. Отклонение высоты от дна пробирки до градуировочной риски, соответствующей 50 мл, у разных пробирок не должно превышать 2 мм.
A.4 Приготовление раствора пробы и контрольного раствора
A.4.1 Расчет количества пробы для испытаний
Используемое количество пробы в граммах рассчитывают по формуле, приведенной ниже. Количество стандартного раствора свинца обычно составляет 2,0 мл, однако может быть изменено с целью достижения желаемого предела и оптимизации условий испытаний для повышения надежности метода.
где w - масса пробы, г;
V - объем стандартного раствора свинца, мл;
L - предельное содержание тяжелых металлов, мкг/г.
A.4.2 Выбор метода подготовки проб
Метод 1 может применяться при исследованиях сырьевых материалов, растворяющихся в воде и не образующих осадка при добавлении разведенной уксусной кислоты для регулирования значения pH раствора в интервале от 3,0 до 3,5. Если данные требования не выполняются или уровень извлечения недостаточен, изучают возможность применения методов со 2 по 4. Если ни один из указанных способов не является приемлемым, используют иные методы, не описанные здесь.
Метод 2 обычно применяется при исследованиях органических материалов. В то же время он неприменим для анализа сырьевых материалов, которые содержат восстанавливающие металлы, такие как медь, и нерастворимые оксиды металлов, образованные в результате озоления, поскольку соляная кислота, применяемая в данном методе, не способна полностью растворить подобные материалы.
Метод 3 часто бывает эффективен при исследованиях сырьевых материалов, полное озоление которых в соответствии с методом 2 не представляется возможным. Он предусматривает растворение имеющихся остатков при помощи царской водки. При подготовке пробы в соответствии с данным методом не следует использовать платиновые тигли.
Метод 4 может применяться, если уровень извлечения, достигнутый в соответствии с методом 2, является недостаточным. Сжигание с этанолом в присутствии магния нитрата до образования золы позволяет предотвратить испарение металлов в процессе горения.
Методы со 2 по 4 предусматривают прокаливание или кислотную минерализацию в открытых емкостях. Ртуть и мышьяк могут испаряться в этом процессе. Соответственно при исследовании этих элементов может потребоваться особый подход.
A.4.3 Метод 1
Необходимое расчетное количество пробы помещают в пробирку Несслера и разводят в достаточном количестве воды до получения объема 40 мл. Добавляют 2 мл разбавленной уксусной кислоты и воду, доводя объем раствора до 50 мл. Полученный раствор используют для испытаний. Соответствующее количество стандартного раствора свинца помещают в другую пробирку Несслера, затем добавляют 2 мл разбавленной уксусной кислоты и воду, доводя объем раствора до 50 мл. Полученный раствор используют в качестве контрольного.
A.4.4 Метод 2
Помещают необходимое расчетное количество пробы в кварцевый или фарфоровый тигель, неплотно закрывают крышкой и обугливают при небольшом нагреве. После охлаждения добавляют 2 мл азотной кислоты и пять капель серной кислоты, осторожно нагревают до исчезновения белой пены, а затем прокаливают при температуре от 450°C до 600°C до озоления. После охлаждения добавляют 2 мл соляной кислоты, выпаривают досуха на водяной бане, увлажняют остатки тремя каплями соляной кислоты, добавляют 10 мл горячей воды и прогревают в течение 2 мин. Добавляют одну каплю титрованного раствора фенолфталеина, а затем по капле добавляют титрованный раствор аммиака вплоть до появления бледно-розового окрашивания. При добавлении избыточного количества раствора аммиака испытуемый раствор может покраснеть, а последующее добавление 2 мл разбавленной уксусной кислоты может не обеспечить получение значения pH, приблизительно равного 3,5, и таким образом точная оценка содержания элементов станет затруднительной. Если количество раствора аммиака все же оказалось избыточным, в испытуемый раствор добавляют разбавленную соляную кислоту для его нейтрализации до тех пор, пока раствор снова не станет бледно-розовым.
Добавляют 2 мл разбавленной уксусной кислоты, при необходимости фильтруют, промывают 10 мл воды, переносят фильтрат и промывную воду в пробирку Несслера, добавляют воду до получения объема 50 мл и используют полученный раствор для испытаний.
Приготавливают контрольный раствор следующим образом: берут 2 мл азотной кислоты, 5 капель серной кислоты и 2 мл соляной кислоты, выпаривают вначале на водяной бане, а затем досуха на песчаной бане и увлажняют остаток тремя каплями соляной кислоты. Выполняют те же операции, что и при приготовлении испытуемого раствора, и добавляют необходимое расчетное количество стандартного раствора свинца и воды до получения объема 50 мл.
A.4.5 Метод 3
Помещают необходимое расчетное количество пробы в кварцевый или фарфоровый тигель. Начинают с медленного нагревания, затем прокаливают до разложения. После охлаждения добавляют 1 мл царской водки, выпаривают досуха на водяной бане, увлажняют остатки тремя каплями соляной кислоты, добавляют 10 мл горячей воды и прогревают в течение 2 мин. Добавляют одну каплю титрованного раствора фенолфталеина, а затем по капле добавляют титрованный раствор аммиака вплоть до появления бледно-розового окрашивания. Добавляют 2 мл разбавленной уксусной кислоты, при необходимости фильтруют, промывают 10 мл воды, переносят фильтрат и смывку в пробирку Несслера, добавляют воду до получения объема 50 мл и используют полученный раствор для испытаний.
Приготавливают контрольный раствор следующим образом: выпаривают досуха 1 мл царской водки на водяной бане, выполняют те же операции, что и при приготовлении испытуемого раствора, а затем добавляют необходимое расчетное количество стандартного раствора свинца и воды до получения объема 50 мл.
A.4.6 Метод 4
Помещают необходимое расчетное количество пробы в платиновый или фарфоровый тигель, добавляют 10 мл раствора магния нитрата гексагидрата в этаноле (1:10) и тщательно перемешивают. Поджигают этанол и обугливают при небольшом нагревании. После охлаждения добавляют 1 мл серной кислоты, осторожно нагревают и прокаливают при температуре от 500°C до 600°C до разложения. Если часть обугленного вещества сохраняется при использовании данного метода, остатки увлажняют небольшим количеством серной кислоты и снова прокаливают до разложения. После охлаждения добавляют к остатку 3 мл соляной кислоты и выпаривают досуха на водяной бане. Увлажняют остаток тремя каплями соляной кислоты, добавляют 10 мл горячей воды и нагревают до растворения. Добавляют одну каплю титрованного раствора фенолфталеина, а затем по капле добавляют титрованный раствор аммиака вплоть до появления бледно-красного окрашивания. Добавляют 2 мл разбавленной уксусной кислоты, при необходимости фильтруют, промывают 10 мл воды и переносят фильтрат и смывку в пробирку Несслера. Добавляют воду, доводя объем раствора до 50 мл, и используют полученный раствор в качестве испытуемого раствора.
Приготавливают контрольный раствор следующим образом: берут 10 мл раствора магния нитрата гексагидрата в этаноле (1:10) и воспламеняют этанол. После охлаждения добавляют 1 мл серной кислоты, осторожно нагревают и прокаливают при температуре от 500°C до 600°C. После охлаждения добавляют 3 мл соляной кислоты, выполняют те же операции, что и при приготовлении испытуемого раствора, и добавляют необходимое расчетное количество стандартного раствора свинца и воды до получения объема 50 мл.
A.5 Определение
Добавляют к испытуемому раствору и контрольному раствору по одной капле титрованного раствора сульфида натрия, тщательно перемешивают и дают отстояться в течение 5 мин. Важно, чтобы раствор сульфида натрия добавлялся к испытуемому и контрольному раствору одновременно, поскольку полнота окрашивания зависит от длительности выдержки. Рассматривают растворы на белом фоне в горизонтальной или вертикальной плоскости и сравнивают их окраску. Окраска испытуемого раствора не должна быть интенсивнее окраски контрольного раствора.
При сравнении окраски растворов особое внимание следует обратить на правильность установки источника света, чтобы избежать ошибочного заключения. Наблюдение выполняют при достаточной яркости освещения. При этом освещенность испытуемого и стандартного растворов должна быть одинаковой. Оптимальное взаимное расположение пробирки Несслера и белого фона, в качестве которого выступает бумага или картон, при визуальном контроле показано на рисунке 1. Перед наблюдением из пробирки Несслера удаляют пробку. Если различие в окраске растворов с уверенностью оценить не удается, меняют положение испытуемого и стандартного растворов и повторяют наблюдение. Этот шаг помогает избежать неверного заключения при визуальном контроле.
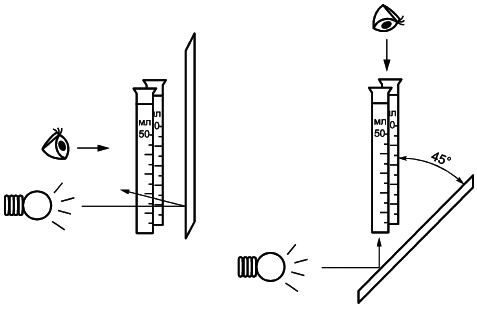 |
Рисунок A.1 - Схема визуального контроля
Приложение B
(справочное)
Рентгеновская флуоресценция
B.1 Общие положения
Элементы, определяемые с использованием данного метода, описаны в 2.4.3.1 и 2.4.3.2.
B.2 Оборудование
B.2.1 Рентгенофлуоресцентный спектрометр с соответствующим детектором.
B.2.2 Оборудование для подготовки проб при необходимости, например пресс-форма для таблеток, кюветы для образцов и тонкая пленка.
B.3 Подготовка пробы
При выполнении анализа порошкообразных проб в случае необходимости к ним может добавляться связующее вещество, чтобы облегчить формирование таблеток прессованием. Для анализа жидких проб их запечатывают в соответствующие емкости с тонкой пленкой. Если проба представлена жидкостью и концентрация измеряемого элемента в ней невелика, фактическая концентрация может быть повышена с применением разнообразных методов концентрирования.
B.4 Детектирование
Если детектор нуждается в охлаждении, его заблаговременно охлаждают жидким азотом перед началом анализа. Устанавливают напряжение рентгеновской трубки, ток рентгеновской трубки, время измерения и другие необходимые параметры, предусмотренные поставщиком оборудования. Выбирают искомые элементы и приступают к анализу.
Отдельные приборы могут обеспечивать работу по методу фундаментальных параметров. В соответствии с этим методом количество элементов в пробе определяется без построения кривой калибровки путем оценки на основе теоретических расчетов с использованием информации о количестве каждого элемента, содержащегося в пробе, для корректировки влияния матрицы. Кроме того, некоторое оборудование предполагает использование коэффициентов преобразования, основанных на том же самом теоретическом уравнении, что и метод фундаментальных параметров. Эти коэффициенты могут использоваться только для упрощенного количественного определения.
Количественное определение обычно выполняется на основе кривой калибровки, которая строится исходя из интенсивности флуоресцентного рентгеновского излучения целевого элемента. Интенсивность рентгеновской флуоресценции зависит либо от характеристик составляющих элементов, в первую очередь основных, либо от размера частиц. Эту зависимость описывают как влияние матрицы. Соответственно для получения линейной кривой калибровки стандартные образцы по своему химическому составу и состоянию поверхности должны быть аналогичны исследуемым пробам.
В дополнение к вышеописанному возможно применение так называемого метода внутреннего стандарта, в соответствии с которым к пробе добавляют элемент с постоянным значением концентрации, выступающий как внутренний стандарт. Значение концентрации определяют на основе отношения интенсивностей для элемента, являющегося внутренним стандартом, и целевого элемента.
Приложение C
(справочное)
Количественное определение элементов в пробах
C.1 Количественное определение с использованием ААС [9], [22]
C.1.1 Общие положения
Для минерализации проб может использоваться микроволновая печь. Определение количества мышьяка, кадмия и свинца на основе ГП-ААС по методу внешней калибровки выполняется с применением соответствующих растворов. Поскольку область применения ААС не ограничена указанными элементами, подобные исследования могут проводиться и для других элементов при соблюдении надлежащих условий подготовки проб.
C.1.2 Реактивы и растворы
C.1.2.1 Общие положения
Все реактивы должны иметь степень чистоты "чистый для анализа". При наличии возможности предпочтительнее использовать реактивы чистые для элементного анализа.
C.1.2.2 Модификатор для мышьяка, раствор палладия концентрацией 1000 мкг/мл в растворе с объемной долей 0,5% азотной кислоты.
C.1.2.3 Модификатор для свинца и кадмия, смесь 1:1 растворов концентрацией 2,0 г/л магния нитрата в растворе с объемной долей 0,5% азотной кислоты и концентрацией 2,0 г/л аммония фосфата одноосновного в растворе с объемной долей 0,5% азотной кислоты.
C.1.2.4 Стандартные растворы, мышьяка, кадмия и свинца концентрацией 1000 мкг/мл для каждого элемента.
C.1.2.5 Градуировочные растворы, стандартные растворы (C.1.2.4) разбавляют раствором с объемной долей азотной кислоты 0,5% для получения градуировочных растворов с различными значениями концентрации. Ее диапазон должен охватывать верхние и нижние значения концентрации целевого элемента.
C.1.3 Оборудование
C.1.3.1 Фильтровальная бумага с размером пор, обеспечивающим удерживание частиц размером 11 мкм.
C.1.3.2 Система микроволновой минерализации с регулируемыми значениями давления и температуры.
C.1.3.3 Емкости из кварца или тетрафторметана (ТФМ) вместимостью 50 мл.
C.1.4 Методика испытаний
C.1.4.1 Подготовка пробы
Тщательно взвешивают две одинаковые навески пробы массой от 0,15 до 0,20 г и помещают в стойкий к высокому давлению сосуд из кварца или тетрафторметана вместимостью 50 мл. Если проба содержит летучие компоненты, эти компоненты следует удалить перед микроволновой минерализацией. Добавляют 3 мл концентрированной азотной кислоты (не менее 65%) и 1 мл раствора перекиси водорода с объемной долей 30% с использованием градуированной пипетки. Если проба содержит тальк или пигменты, добавляют 1 мл концентрированной соляной кислоты. Убеждаются, что проба полностью увлажнилась. Закрывают крышку емкости. Дают отстояться в течение не менее 15 мин в зависимости от типа матрицы для завершения реакции. Приступают к обработке пробы в системе микроволновой минерализации. Задают настройки микроволновой минерализации и запускают выполнение программы. Для достижения приемлемого уровня извлечения условия микроволновой минерализации должны быть оптимизированы для конкретных видов проб. При необходимости обращаются за консультацией к изготовителю системы микроволновой минерализации:
- максимальная температура - 200°C;
- максимальное давление - 7,5 МПа;
- продолжительность минерализации - от 40 до 50 мин в зависимости от пробы.
Если прецизионность полученных результатов, степень извлечения или оба этих показателя представляются неудовлетворительными, рекомендуется изменить либо количество пробы, либо количество применяемых для минерализации кислот, либо внести изменения в программу микроволновой минерализации.
После минерализации пробу охлаждают при комнатной температуре, добавляют 20 мл деионизированной воды к минерализованному раствору, ополаскивают наружные стенки емкости и плотно закрывают крышкой. Фильтруют через фильтровальную бумагу, помещают в мерную колбу вместимостью 50 мл и разбавляют до необходимого объема деионизированной водой.
Приготавливают холостой раствор для проверки метода таким же образом, как и предыдущий раствор, но без добавления пробы.
C.1.4.2 Проба с добавками для определения процентной доли извлечения
Тщательно взвешивают с точностью до 0,1 мг две одинаковые навески пробы массой от 0,15 до 0,20 г и помещают в стойкий к высокому давлению сосуд из кварца или тетрафторметана вместимостью 50 мл. Пипеткой помещают в емкость по 300 мкл стандартного раствора свинца с концентрацией 5 мкг/мл, стандартного раствора мышьяка с концентрацией 5 мкг/мл и стандартного раствора кадмия с концентрацией 0,5 мкг/мл. Минерализуют для получения раствора пробы, как описано в C.1.4.1.
C.1.4.3 Детектирование
В таблице 1 приведен пример условий, обеспечиваемых для ГП-ААС. Задают соответствующие параметры программы анализа. При запуске последовательности операций следуют требованиям руководства по эксплуатации, предоставленного поставщиком оборудования.
Таблица C.1 - Условия применения ГП-ААС
Элемент | Длина волны, нм | Пиролиз, °C | Температура атомизации, °C | Объем модификатора, мкл
| Вводимый объем, мкл |
Мышьяк | 193,7 | 1250 | 2100 | 5 | 20 |
Кадмий | 228,8 | 650 | 1650 | 5 | 20 |
Свинец | 283,3 | 550 | 1550 | 5 | 20 |
С.1.5 Расчеты
Линейная зависимость между возрастающей концентрацией рассматриваемого элемента в градуировочных растворах и соответствующей площадью пика представлена формулой (С.1):
где а - наклон линии регрессии;
b - отрезок по оси Y.
Используют значения наклона и размера отрезка, полученные для градуировочных растворов, и показания оборудования для испытуемого раствора, чтобы рассчитать концентрацию элемента в испытуемом растворе в микрограммах на грамм. Количество искомого элемента в пробе вычисляют по следующим формулам:
А - показания оборудования при анализе раствора пробы, интенсивность;
b - отрезок по оси Y;
а - наклон кривой калибровки;
m - масса исследуемой пробы, г;
V - составляет 50 мл;
w - количество искомого элемента в пробе, мкг/г.
С.2 Количественное определение с использованием ИСП
С.2.1 Общие положения
Данный метод описывает порядок количественного определения свинца, кадмия, мышьяка, никеля, кобальта, хрома в разнообразных матрицах. Поскольку область применения ИСП не ограничена указанными элементами, подобные исследования могут проводиться и для других элементов при соблюдении надлежащих условий подготовки проб. При использовании ИСП могут использоваться различные режимы детектирования, такие как ОЭС или МС. Далее в качестве примера рассматривается применение ИСП-МС.
C.2.2 Реактивы и растворы
Для анализа ИСП-МС используют реактивы элементов соответствующей степени чистоты.
C.2.3 Стандартные растворы
C.2.3.1 Многоэлементный стандартный раствор, содержащий свинец, кадмий, мышьяк, никель, кобальт и хром в концентрации 10 мкг/мл, или стандартные растворы элементов свинца, кадмия, мышьяка, никеля, кобальта и хрома с концентрацией 10 мкг/мл.
C.2.3.2 Индий для ИСП-МС (раствор), 1000 мкг/мл (внутренний стандарт 1, по выбору).
C.2.3.3 Родий для ИСП-МС (раствор), 1000 мкг/мл (внутренний стандарт 2, по выбору).
C.2.4 Оборудование
C.2.4.1 Система микроволновой минерализации, приспособленная для подготовки проб при использовании ИСП.
C.2.4.3 Аналитические весы с точностью измерения до 0,1 мг.
C.2.4.4 Мерная полипропиленовая колба вместимостью 50 мл.
C.2.4.5 Шприцевые фильтры, 25 мм, с диаметром пор 0,45 мкм с гидрофильным полиэфирсульфоном или аналогичные.
C.2.4.6 Одноразовые шприцы с измеряемым объемом до 5 мл.
C.2.5 Приготовление стандартных растворов
C.2.5.1 Разбавляющий раствор: в полипропиленовой колбе вместимостью 500 мл смешивают 250 мл очищенной воды, 50 мл концентрированной азотной кислоты (не менее 65%) и 10 мл раствора перекиси водорода с объемной долей 30%. Доливают очищенную воду, чтобы довести объем раствора до 500 мл. Закрывают колбу и перемешивают. Раствор приготавливают перед каждым анализом.
C.2.5.2 Стандартный раствор: помещают в полипропиленовую мерную колбу вместимостью 100 мл 1,0 мл многоэлементного раствора (10 мкг/мл). Доводят объем раствора до 100 мл разбавляющим раствором и хорошо перемешивают. Эту процедуру повторяют не реже одного раза в месяц в зависимости от регулярности проведения анализов. Конечное значение концентрации каждого элемента должно составлять 100 нг/мл.
C.2.5.3 Растворы для построения кривой калибровки: растворы для построения кривой калибровки приготавливают с применением стандартного раствора с концентрацией 100 нг/мл. Стандартная кривая строится в диапазоне концентраций от 0,1 до 100 нг/мл. Ее диапазон должен охватывать верхние и нижние значения концентрации целевого элемента. Контроль качества выполняется со значением концентрации 10 нг/мл.
C.2.5.4 Раствор внутреннего стандарта: в полипропиленовую колбу вместимостью 1000 мл помещают 500 мл очищенной воды, 100 мл концентрированной азотной кислоты и 20 мл раствора перекиси водорода с объемной долей 30%. Добавляют 1 мл раствора индия (C.2.3.2) и 1 мл раствора родия (C.2.3.3). Доливая очищенную воду, доводят объем раствора до 1000 мл. Закрывают колбу и перемешивают. Эту процедуру повторяют не реже одного раза в месяц в зависимости от регулярности проведения анализов. Конечное значение концентрации каждого элемента должно составлять 1 мкг/мл.
C.2.6 Подготовка пробы
В процессе приготовления проб для их минерализации перед измерением с помощью оборудования для ИСП-МС могут применяться обработка смесью концентрированной азотной кислоты и раствора перекиси водорода с объемной долей 30%, а также микроволновая обработка. Пробы могут приготавливаться и без микроволновой обработки - в этом случае минерализацию необходимо выполнять в закрытой емкости. С основной информацией о порядке минерализации проб в закрытых сосудах можно ознакомиться в [23].
Тщательно взвешивают не менее 0,2 г пробы и помещают ее в соответствующую емкость для микроволновой минерализации. Добавляют 5 мл концентрированной азотной кислоты, 1,5 мл очищенной воды и 1 мл раствора перекиси водорода с концентрацией 30% объемных долей. Таким же образом приготавливают раствор холостой пробы.
Задают настройки микроволновой минерализации и запускают выполнение программы. Для достижения приемлемого уровня извлечения условия микроволновой минерализации должны быть оптимизированы для конкретных видов проб. После минерализации растворы проб охлаждают до комнатной температуры. Переносят раствор пробы из емкости, в которой выполнялась минерализация, в полипропиленовую мерную колбу вместимостью 50 мл. Ополаскивают емкость трижды водой ультравысокой чистоты* и переносят промывную воду в ту же колбу. Разбавляют водой ультравысокой очистки до получения окончательного объема 50 мл. Тщательно перемешивают полученный раствор. Немедленно отфильтровывают 5 мл гидролизата с использованием фильтра с размером пор 0,45 мкм и помещают в виалу для ИСП-МС.
_________________
* На территории Республики Беларусь вместо воды ультравысокой чистоты допускается применять воду первой степени чистоты по ГОСТ ISO 3696-2013.
C.2.7 Выполнение анализа методом ИСП-МС
Каждый раз перед проведением нового исследования выполняют настройку и проверку оборудования для ИСП-МС (доля двухзарядных ионов, оксидов, эффективность коллизионной ячейки, а также чувствительность). В таблице 2 показаны отношение масса/заряд и режим анализа для каждого элемента. Определяют необходимые условия для ИСП-МС в соответствии с руководством по эксплуатации, предоставленным поставщиком оборудования.
Таблица C.2 - Масса каждого элемента и соответствующий режим
Элемент | Отношение массы к заряду, масса/заряд
| Режим |
Хром | 52/53 | He |
Кобальт | 59 | He |
Никель | 60/62 | He |
Мышьяк | 75 | He |
Кадмий | 111/114 | He/без газа |
Свинец | 206/207/208 | Без газа |
C.2.8 Анализ проб
Определяют последовательность операций в соответствии с руководством по эксплуатации и приступают к анализу пробы.
С.2.9 Расчеты
Линейная зависимость между возрастающей концентрацией рассматриваемого элемента в градуировочных растворах и соответствующей площадью пика представлена формулой (С.4):
где а - наклон линии регрессии;
b - отрезок по оси Y.
Используют значения наклона и размера отрезка, полученные для градуировочных растворов, и показания оборудования для испытуемого раствора, чтобы рассчитать концентрацию элемента в испытуемом растворе в микрограммах на грамм. Количество искомого элемента в пробе вычисляют по следующим формулам:
А - показания оборудования при анализе раствора пробы, интенсивность;
b - отрезок по оси Y;
а - наклон кривой калибровки;
m - масса исследуемой пробы, г;
V - составляет 50 мл;
w - количество искомого элемента в пробе, мкг/г.
Библиография
[1] | Al-Saley S. Al-Enazi, N. Shinwari, Assessment of lead in cosmetic products. Regul. Toxicol. Pharmacol. 2009, 54 pp.105-113 (Оценка содержания ртути в косметической продукции) | |
[2] | Besecker K. et al. A simple closed-vessel nitric acid digestion method for cosmetic samples. Atomic Spectroscopy. 1998, 19 pp.48-54 (Упрощенный метод минерализации в закрытом сосуде с азотной кислотой для проб косметической продукции) | |
[3] | YAKUJI NIPPO. Ltd. The Japanese Standards of Quasi-drug Ingredients, 2006 (Японские стандарты "Ингредиенты квазинаркотики") | |
[4] | European Directorate for Quality of Medicines & HealthCare. European Pharmacopoeia. ISBN/ISSN, Seventh Edition, 2011 (Европейский директорат по качеству лекарственных средств для здравоохранения. Европейская фармакопея) | |
[5] | YAKUJI NIPPO. Ltd. The Japanese Standards of Cosmetic Ingredients. Newly, revised edition, 20 (Японские стандарты по косметическим ингредиентам. Новое, переработанное издание) | |
[6] | YAKUJI NIPPO, Ltd., The Japanese Cosmetic Ingredients Codex 1993. 1994 (Японский кодекс по косметическим ингредиентам) | |
[7] | U.S. Pharmacopeial Convention, USP35-NF 30, 2012-11-29 (Фармакопейная конвенция США) | |
[8] | Gernez G., & Moine I. sur l’essai des lourds. Pharmeuropa, 1989. 1, 249-251 (5) (Об испытаниях на наличие тяжелых металлов) | |
[9] | INDIAN STANDARD PCD 19(2698) Methods of test for cosmetics - Determination of heavy metals (Arsenic, Cadmium, Lead and Mercury) by atomic absorption spectrometry (AAS), March 2013 (Draft document) (Определение содержания тяжелых металлов (мышьяк, кадмий, свинец и ртуть) методом атомно-абсорбционной спектрометрии (ААС), март 2013 г. (проект)) | |
[10] | Torres D.P., Martins-Teixeira M.B., Silva E.F., Queiroz H.M. Method development for the control determination of mercury in seafood by solid-sampling thermal decomposition amalgamation atomic absorption spectrometry (TDA AAS) Food Additives and Contaminants, 29, 4, April 2012, 625-632 (Разработка метода для контроля содержания ртути в морепродуктах средствами атомно-абсорбционной спектрометрии с термическим разложением и амальгамацией твердых проб (ТРА ААС) | |
[11] | UK Environmental Agency, The determination of metals in solid environmental samples. Blue Book 204, 2006 (Определение содержания металлов в твердых пробах, взятых из окружающей среды) | |
[12] | CEN § 64 LFGB K 84.00-29, Druckaufschluss zur Bestimmung von Elementen in kosmetischen Mitteln. 2011 (Минерализация под давлением для определения элементов в косметической продукции) | |
[13] | Hepp N.M., Mindak W.R., Cheng J. Determination of total lead in lipstick: Development and validation of a microwave-assisted digestion, inductively coupled plasma-mass spectrometric method. J. Cosmet. Sci. 2009, 60 pp.405-414 (Определение общего содержания свинца в губной помаде. Разработка и валидация метода масс-спектрометрии с индуктивно связанной плазмой с применением микроволновой минерализации) | |
[14] | Beckhoff B., Kanngiesser B., Langhoff N., Wedell R., Wolff H. Handbook of Practical X-Ray Fluorescence Analysis. Springer, 2006 (Практическое руководство по рентгенофлуоресцентному анализу) | |
[15] | Welz B. et al. Atomic Absorption Spectrometry. Wiley, 1999 (Атомно-абсорбционная спектрометрия) | |
[16] | Butala Steven J.M., Scanlan Larry P., Chaudhuri Sanwat N. A Detailed Study of Thermal Decomposition, Amalgamation/Atomic Absorption Spectrophotometry Methodology for the Quantitative Analysis of Mercury in Fish and Hair. Journal of Food Protection, 69, 11, November 2006, 2720-2728 (9) (Подробное исследование термического разложения и амальгамации в атомно-абсорбционной спектрофотометрии для количественного анализа содержания ртути в рыбе и волосах) | |
[17] | Ministry of the Environment. Mercury Analysis Manual, Japan, 2004 (Руководство по анализу ртути) | |
[18] | Beauchemin D. Inductively Coupled Plasma Mass Spectrometry. Anal. Chem. 2008, 80 pp.4455-4486 (Масс-спектрометрия с индуктивно связанной плазмой) | |
[19] | Nelms Simon Inductively coupled plasma mass spectrometry handbook. Blackwell, 2005. ISBN-10 0849323819 (Руководство по масс-спектрометрии с индуктивно связанной плазмой) | |
[20] | EN 15763:2009 | Foodstuffs. Determination of trace elements. Determination of Arsenic, Cadmium, mercury and lead in foodstuffs by ICP/MS after pressure digestion (Продукты пищевые. Определение следовых элементов. Определение мышьяка, кадмия, ртути и свинца в пищевых продуктах по методу ИСП/МС после минерализации под давлением) |
[21] | Nolte J. ICP Emission Spectrometry A Practical Guide. Wiley, 2003 (Эмиссионная спектрометрия. Практическое руководство) | |
[22] | ACM THA 05, No. 2, 26/03/2013 (the version presented at ACTLC meeting in Laos on 21/05/2013), Determination of heavy metals (arsenic, cadmium, lead and mercury) in cosmetic products (Определение тяжелых металлов (мышьяка, кадмия, свинца и ртути) в косметической продукции) | |
[23] | U.S. Pharmacopeial Convention, USP36-NF31 (233) - Elemental impurities - Procedures. 2013 (Микролементные загрязнения. Методики анализа) | |
УДК 665.58.012:549.2.06(083.74)(476) | МКС 71.100.70
| IDT |
Ключевые слова: продукция парфюмерно-косметическая, метод скрининга, количественное определение, тяжелые металлы | ||
