
БЗ 11-96

ГОСУДАРСТВЕННЫЙ СТАНДАРТ
СОЮЗА ССР
ПРОДУКТЫ ЛЕСОХИМИЧЕСКИЕ
ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД АНАЛИЗА
ГОСТ 21533-76
Издание официальное
ИПК ИЗДАТЕЛЬСТВО СТАНДАРТОВ Москва
УДК 634.0.86.001.4:006.354 Группа Л49
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
ПРОДУКТЫ ЛЕСОХИМИЧЕСКИЕ
Газохроматографический метод анализа
Wood chemical products. Method of test by gas chromatography
ГОСТ
21533-76
ОКСТУ 2409
Дата введения 01.01.77
Настоящий стандарт распространяется на лесохимические продукты и устанавливает газохроматографический метод определения в них массовой доли основного вещества и примесей.
(Измененная редакция, Изм. Ns 1).
1. АППАРАТУРА, РЕАКТИВЫ И ПОСУДА
1.1. Для проведения анализа применяют следующую аппаратуру, реактивы и посуду: хроматограф газовый с детектором по теплопроводности с чувствительностью по пропану в гелии не менее 110“2 % по объему или детектором ионизации в пламени с чувствительностью по пропану не менее 110~6 % по объему:
носители твердые: хроматон N, хроматон N-AW или диатомито-вый кирпич теплоизоляционный по ГОСТ 2694 размером частиц 0,15—0,2; 0,2—0,25 или 0,25—0,32 мм, для анализа скипидара и камфена может быть использован также сферохром-1 с тем же размером частиц;
сорбент полисорб-1, размер зерен 0,10—0,25 мм; фазы жидкие неподвижные: трикрезилфосфат, смесь полиэтиленгликоля 400 с апиезоном L или апиезоном N, взятых в соотношении 2:3 или 3:2;
Издание официальное Перепечатка воспрещена
© Издательство стандартов, 1976 © И ПК Издательство стандартов, 1997 Переиздание с Изменениями
газы-носители: гелий технический при использовании детектора по теплопроводности;
азот газообразный технический по ГОСТ 9293 при использовании детектора ионизации в пламени;
водород технический марки А по ГОСТ 3022 для питания детектора ионизации в пламени;
вещества-эталоны: толуол по ГОСТ 5789, ч.д.а. для анализа эти-лацетата;
ундекан, хроматографически чистый, для анализа бутилацетата; растворители: ацетон по ГОСТ 2603, ч. или ч.д.а. хлороформ; бутилацетат по ГОСТ 22300, ч., или бутилацетат технический по ГОСТ 8981, марка А;
вода дистиллированная по ГОСТ 6709;
кислота соляная по ГОСТ 3118, разбавленная 1:1 и концентрированная;
натрий углекислый по ГОСТ 83, раствор с массовой долей 5 %; этилацетат по ГОСТ 22300, ч. или этилацетат технический по ГОСТ 8981, марка А;
бумага универсальная индикаторная; баня водяная;
печь муфельная, обеспечивающая температуру нагрева до 950— 1000 °С, с термопарой типа ТПП или милливольтметром по ГОСТ 9736;
шкаф сушильный, обеспечивающий температуру нагрева до 200 °С;
набор сит «физприбор»; насос водоструйный по ГОСТ 25336; стекловата или металлическая сетка; колба коническая Кн-2—500—40(50) по ГОСТ 25336; колба коническая Кн-1-50-14/23(19/23; 24/29) по ГОСТ 25336 или стаканчик для взвешивания по ГОСТ 25336 или герметично закрывающаяся склянка;
колба перегонная по ГОСТ 25336, вместимостью 250 см3;
кран соединительный стеклянный по ГОСТ 7995, трехходовой;
микрошприц вместимостью 10 мм3;
пипетка 6(7)—2—10 по ГОСТ 20292;
стакан В(11)-1(2)—100 (250) ТХС по ГОСТ 25336;
чашка выпарительная 5 по ГОСТ 9147;
линейка измерительная металлическая по ГОСТ 427;
лупа измерительная общего назначения по ГОСТ 25706 с десятикратным увеличением или микроскоп отсчетный с аналогичным увеличением; интегратор электронный;
весы лабораторные с наибольшим пределом взвешивания 200 г. Допускается применять другие средства измерения с метрологическими характеристиками и оборудование с техническими характеристиками не хуже, а также реактивы по качеству не ниже указанных в настоящем стандарте;
полидиэтиленгликольсукцинат (ПДЭГС); канифоль живичная;
кислота маргариновая, ч., или кислота стеариновая по ГОСТ 9419, ч.д.а.;
бензол по ГОСТ 5955, ч. или х.ч.; спирт этиловый по ГОСТ 18300;
тетраметиламмония гидроокись, раствор с массовой долей 3 или 10 %; хроматон N-AW-DMCS или инертон AW-DMCS, размер зерен 0,16—0,20 мм или 0,20—0,25 мм;
фенолфталеин, водно-спиртовой раствор 3:7 с массовой долей 0,1 %;
цилиндр по ГОСТ 1770 вместимостью 25 см3;
воронка делительная типа ВД по ГОСТ 25336 вместимостью 100
или 250 см3;
бюретка вместимостью 10 или 25 см3;
колба с тубусом по ГОСТ 25336 исполнения 1, 2 вместимостью 500 см3;
воронка Бюхнера по ГОСТ 9147 № 3 или 4; фильтры обеззоленные «синяя лента».
(Измененная редакция, Изм. № 4).
2. ПОДГОТОВКА К АНАЛИЗУ
2.1. Приготовление насадки
Необходимое для заполнения колонки количество твердого носителя взвешивают (результат записывают с точностью до четвертого десятичного знака) и помещают в перегонную колбу, соединенную с водоструйным насосом через трехходовой кран. Необходимое количество неподвижной жидкой фазы, указанное в соответствующих разделах настоящего стандарта, взвешивают (результат записывают с точностью до четвертого десятичного знака), растворяют в соответствующем растворителе и переносят в ту же перегонную колбу. Объем растворителя должен быть таким, чтобы весь твердый носитель был
покрыт раствором жидкой фазы. Перегонную колбу погружают в водяную баню, нагретую до (50±10) °С, и при разрежении 4'104—5* 104 Па (300—400 мм рт.ст.) испаряют растворитель до сухого состояния насадки, осторожно встряхивая содержимое. После этого насадку высушивают в сушильном шкафу при (100±5) °С в течение 2—3 ч,
(Измененная редакция, Изм. № 1, 2).
2.2. Подготовка диатомитового кирпича
При применении в качестве твердого носителя диатомитового кирпича перед нанесением на него неподвижной жидкой фазы в зависимости от природы анализируемых веществ его предварительно обрабатывают соответствующим образом.
Для разделения кислородосодержащих соединений алифатического ряда (этилацетата и бутилацетата) около 200 г раздробленного и просеянного диатомитового кирпича помещают в коническую колбу вместимостью 500 см3 и отмывают водой от пыли до тех пор, пока вода не станет прозрачной, затем доливают раствор соляной кислоты, разбавленной 1:1, полностью покрывая диатомитовый кирпич, и кипятят в течение 6—8 ч, периодически сливая отработанную кислоту и заливая свежие порции такого же объема. Кислоту меняют четыре-пять раз. Затем обработанный кирпич промывают дистиллированной водой до нейтральной реакции по индикаторной бумаге, высушивают в сушильном шкафу при 150—200 °С и в течение 5—6 ч прокаливают в муфельной печи при 900—1000 °С.
Для разделения соединений терпенового ряда диатомитовый кирпич готовят, как указано выше, при этом перед высушиванием его заливают раствором углекислого натрия до полного смачивания твердого носителя, избыток раствора сливают, а обработанный кирпич сначала высушивают при 150—200 *С, а затем прокаливают в муфельной печи при 800 °С в течение 3 ч.
(Измененная редакция, Изм. № 2, 4).
2.3. Заполнение хроматографических колонок
V-образные колонки, предварительно промытые органическими
растворителями, заполняют через воронку приготовленной насадкой при постукивании по колонке деревянной палочкой, концы колонки после заполнения закрывают тампонами из стеклянной ваты или металлической сеткой.
Спиральные или W-образные колонки заполняют насадкой при помощи сжатою воздуха или вакуумного насоса, как показано на черт. 1.
я1”*'
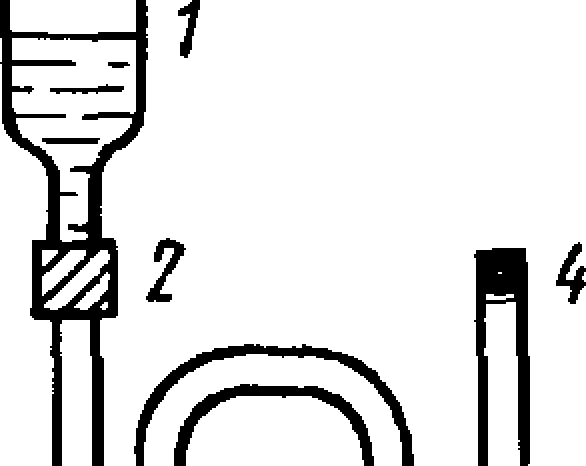
3
Л банцум'насосу
] — стеклянный сосуд для заполнения колонки насадкой; 2~ переходный шланг; 3 —хроматографическая колонка; 4 — уплотнение из стекловаты
Черт. 1
Заполненную колонку закрепляют в термостате хроматографа и, не присоединяя к детектору, продувают газом-носителем со скоростью 5—10 см3/мин при температуре на 5—10 °С ниже максимальной рабочей температуры неподвижной жидкой фазы в течение 20—30 ч. Затем колонку подключают к детектору и устанавливают стабильную нулевую линию при максимальной чувствительности прибора.
(Измененная редакция, Изм. № 2, 3).
2.4. Монтаж, наладку и вывод хроматографа на рабочий режим проводят в соответствии с инструкцией, прилагаемой к прибору.
2.5. Градуировка прибора
Градуировку прибора проводят по искусственным смесям. Их готовят из анализируемого компонента и вещества-эталона, взятых в соотношении 1:1, 2:1, 3:1. Анализируемый компонент и эталон взвешивают (результат записывают с точностью до четвертого десятичного знака) в плотно закрытой конической колбе вместимостью 50 см3.
Каждую искусственную смесь хроматографируют не менее пяти раз.
При использовании метода внутренней нормализации в качестве вещества-эталона используют один из компонентов анализируемого продукта. Относительный поправочный коэффициент {К) для каждого определяемого компонента вычисляют по формуле
где S{ — площадь пика определяемого компонента, мм2;
S3T — площадь пика вещества-эталона, мм2;
тг — масса определяемого компонента, г;
тЭТ — масса вещества-эталона, г.
Поправочный коэффициент вещества-эталона равен единице.
(Измененная редакция, Изм. № 2)*
2.6. Построение градуировочных графиков или нахождение градуировочных коэффициентов для определения массовой доли воды в этила цетате и бутил ацетате.
Для построения градуировочных графиков готовят не менее десяти градуировочных смесей с точно известной массовой долей добавленной воды. Массовую долю воды изменяют в пределах 0,01—0,20 % массы пробы.
Смесь готовят в стаканчике для взвешивания в следующей последовательности: взвешивают пустой стаканчик, помещают в него микрошприцем 1—20 мм3 воды, снова взвешивают, добавляют из пипетки 10 см3 бутилацетата или этилацетата и снова взвешивают (результаты записывают до четвертого десятичного знака).
Исходный бутилацетат или этилацетат и градуировочные смеси хроматографируют при условиях, изложенных в п. 5.5.2. Исходный продукт и каждую смесь хроматографируют не менее шести раз.
Находят среднее арифметическое значение высоты пика воды в исходном продукте и исходных смесях и строят градуировочный график в координатах: разность средних высот пиков в миллиметрах в исходном продукте и градуировочных массах — массовая доля добавленной воды в процентах.
Допускается определять градуировочный коэффициент (К) по формуле
п
m h ’
где п — количество градуировочных смесей;
т — массовая доля воды, добавленной в градуировочную смесь,
%;
h — средняя разность между высотой пика воды в градуировочной смеси и исходном продукте, мм.
Градуировочные графики или градуировочные коэффициенты проверяют один раз в два месяца и при замене хроматографических колонок.
(Введен дополнительно, Изм. № 3).
3. ОБЩИЕ ТРЕБОВАНИЯ К ПРОВЕДЕНИЮ АНАЛИЗА
3.1. Пуск и вывод прибора на рабочий режим проводят в соответствии с инструкцией, прилагаемой к прибору.
Режим работы хроматографа указан в соответствующих разделах настоящего стандарта.
3.2. Пробы жидких продуктов вводят в хроматограф непосредственно, а твердых — предварительно растворив в соответствующем растворителе.
При определении примесей методом внутреннего эталона к пробе анализируемого продукта добавляют вещество-эталон. Анализируемый продукт и вещество-эталон взвешивают (результат записывают с точностью до четвертого десятичного знака) в плотно закрытой конической колбе вместимостью 50 см3.
(Измененная редакция, Изм. № 2).
3.3. Пробу анализируемого продукта вводят в колонку при помощи микрошприца через головку испарителя, прокалывая резиновую мембрану, иглу вводят на полную длину и быстро впрыскивают пробу.
4. ОБРАБОТКА РЕЗУЛЬТАТОВ
4.1. Массовую долю каждого компонента анализируемого продукта, определяемого методом внутреннего эталона, (X) в процентах вычисляют по формуле
S.-K.-m 100
__ / I эт
S * т
эт
где т — масса пробы, г;
/иэт — масса вещества-эталона, г;
Sj — площадь пика определяемого компонента, мм2;
5ЭТ — площадь пика вещества-эталона, мм2;
Kt — относительный поправочный коэффициент определяемого компонента.
4.2. Массовую долю каждого компонента анализируемого продукта, определяемого методом внутренней нормализации, (Хх) в процентах вычисляют по формуле
у loo
Al ZSrK, ’
где S{ — площадь пика определяемого компонента, мм2;
Kt — относительный поправочный коэффициент определяемого компонента;
L5, А, — сумма произведений площадей пиков на соответствующие поправочные коэффициенты всех компонентов анализируемого продукта, мм2.
4.1, 4.2. (Измененная редакция, Изм. № 1).
4.2а. Массовую долю воды (Х2) в процентах в бутилацетате или этилацетате вычисляют по градуировочному графику, измерив высоту пика воды, или по формуле
Х2 = К. А,
где К — градуировочный коэффициент;
А — высота пика, мм.
(Введен дополнительно, Изм. № 3).
4.3. Площадь пика каждого компонента анализируемого продукта (Sg) в квадратных миллиметрах вычисляют по формуле
S, = А, • bj ■ А/,.,
где Л, — высота пика, мм;
Ь, — ширина пика, измеренная на половине его высоты, мм;
Mi — масштаб записи сигнала пика.
В расчет принимают ширину линии, очерчивающей пик (черт. 2,а).
При неполном разделении пиков (черт. 2,6) за ширину пика
Ь,
принимают удвоенную полуширину пика у , которая измеряется на половине высоты.
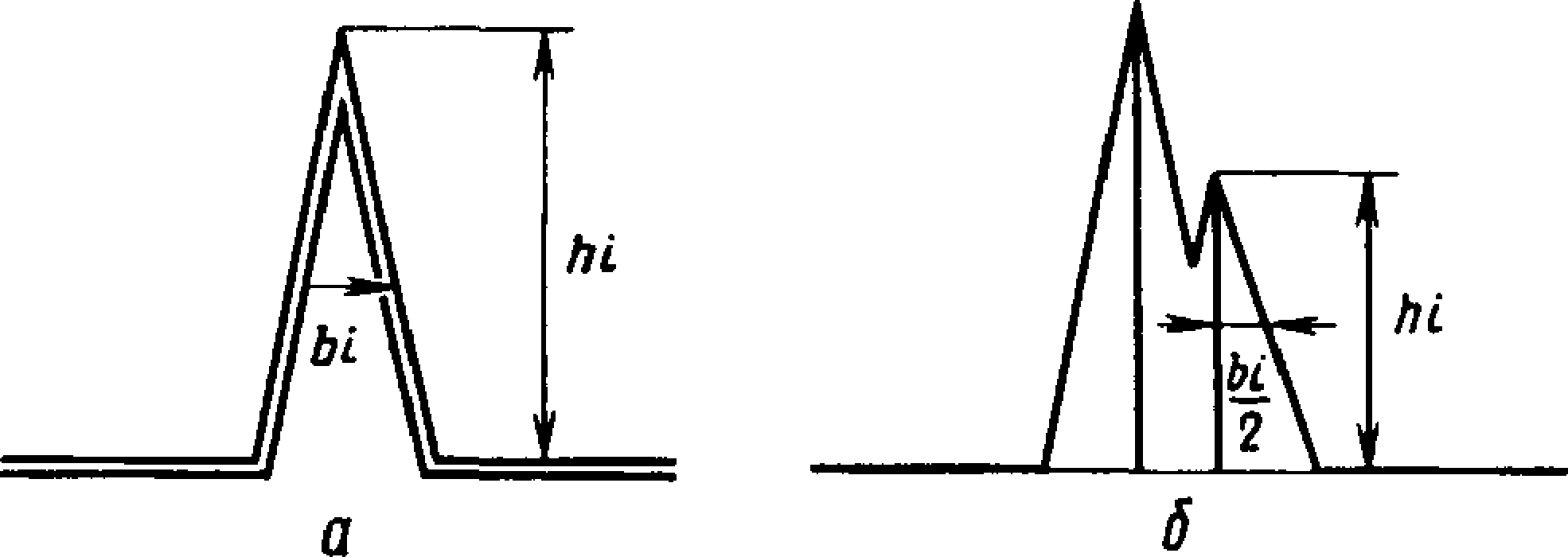
Черт. 2
Площади пиков могут быть измерены в единицах счета интегратора.
(Измененная редакция, Изм. № 2).
5. ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ОСНОВНОГО ВЕЩЕСТВА И ПРИМЕСЕЙ В ТЕХНИЧЕСКОМ ЭТИЛАЦЕТАТЕ И БУТИЛАЦЕТАТЕ
5.1. Сущность метода заключается в газохроматографическом разделении продуктов в насадочной колонке и расчете массовой доли компонентов методом внутреннего эталона. Метод позволяет определять массовую долю компонентов до 0,05 %.
(Измененная редакция, Изм. № 1).
5.2. Три крези л фосфат наносят на твердый носитель в виде раствора в ацетоне.
5.3. Хроматограммы снимают при следующем режиме:
Количество трикрезилфосфата от массы твердого носителя, % | для этил а-цетата 20 | для бутил-ацетата 20-25 |
Длина колонки, м | 2 | 2 |
Внутренний диаметр колонки, мм | 3-4 | 3-4 |
Температура термостата колонки, РС | 65-70 | 88 |
Температура испарителя, *С | 100-125 | 150-200 |
Расход гелия или азота, см3/мин | 20-40 | 25-40 |
Чувствительность потенциометра при использовании детектора по теплопроводности, мВ, не более | 5 | 5 |
Масса навески анализируемого продукта, г | 1-2 | 1-2 |
Количество внутреннего эталона от массы навески, % | 1,5-2 | 1,5-2 |
Объем пробы, вводимой в хроматограф, мм3 | 0,5-5 | 0,5-5 |
Продолжительность одного определения, мин | 25-35 | 25-35 |
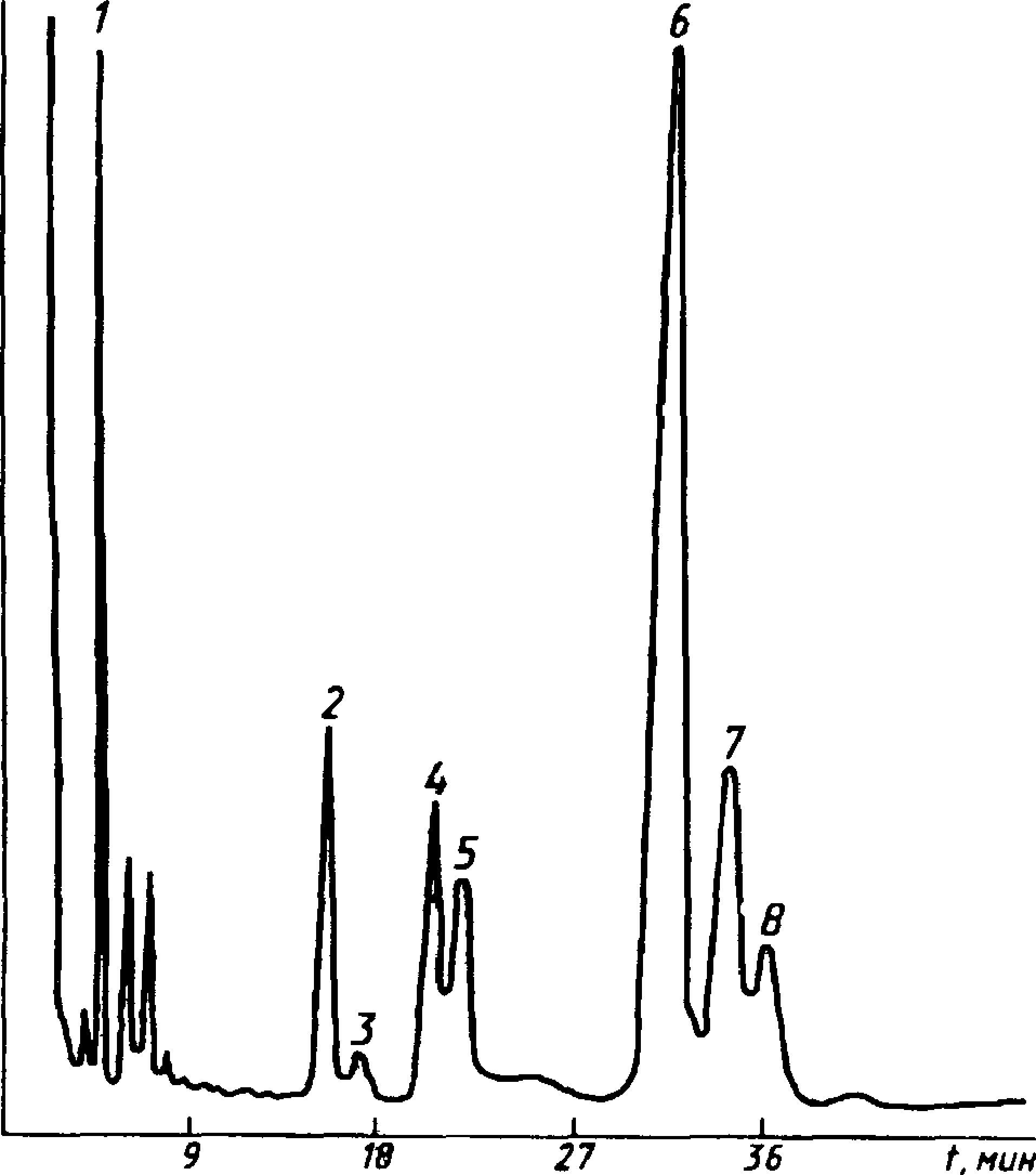
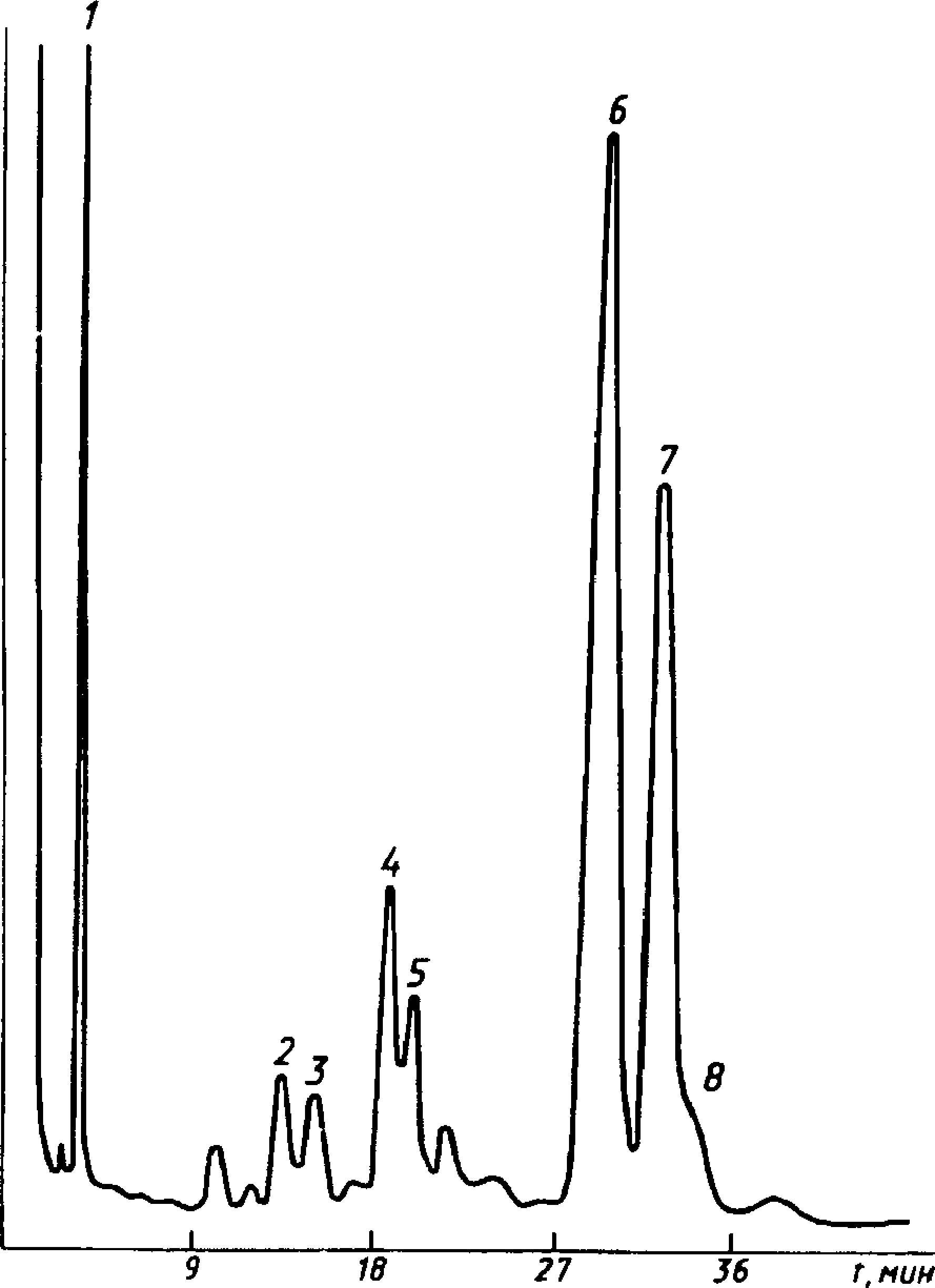
Типовые хроматограммы технического этилацетата и бутилацетата показаны на черт. 3 и 4.
Типовая хроматограмма технического этилацетата
Типовая хроматограмма технического бутилацетата

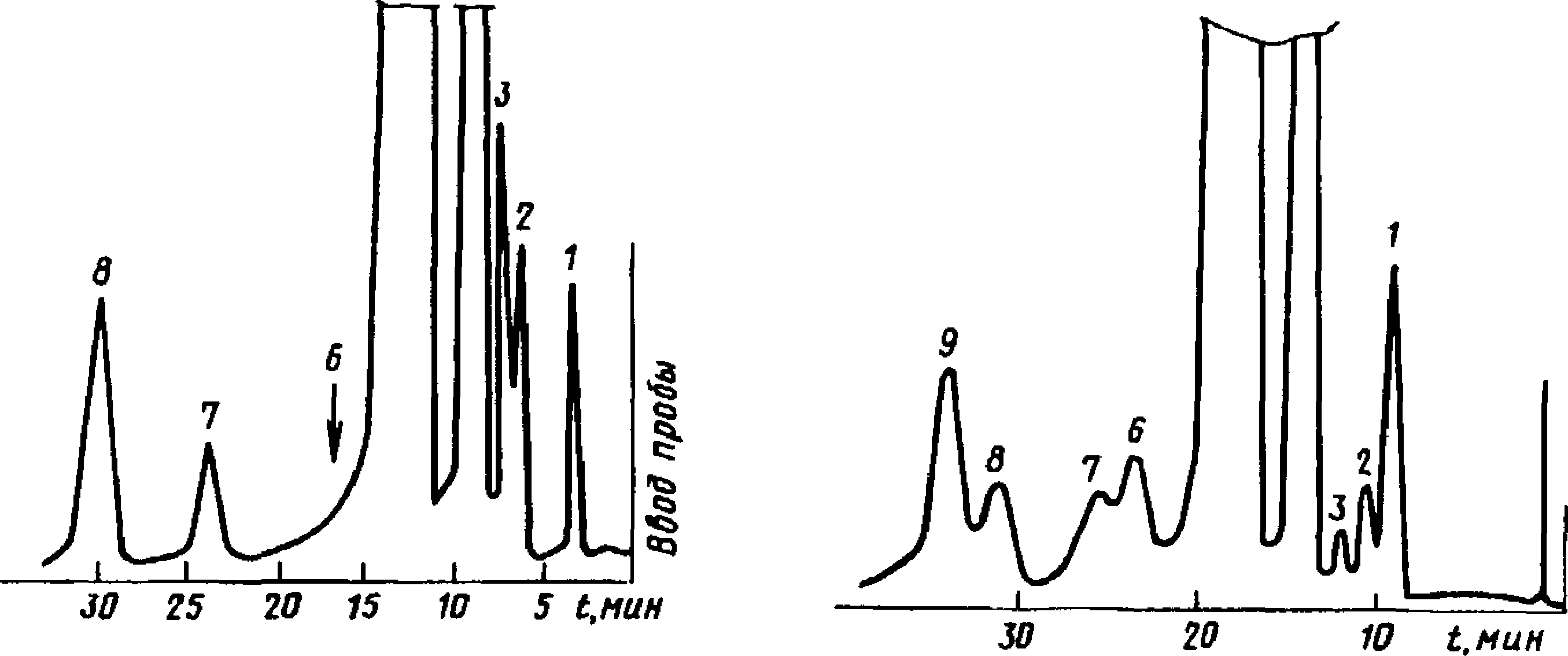
1 — диэтиловый эфир; 2 — этил-формиат; 3 — метилацетат; 4 — этиловый спирт; 5 — этилацетат; 6 — метилпропионат; 7 — этилпропио-нат; 8 — толуол (внутренний эталон)
1 — пропил ацетат; 2 — бутил формиат; 3 — изобутилацетат; 4 — бутиловый спирт; 5 — бутил ацетат; 6 — неидентифицирован-
ный компонент; 7 — изоамилацетат; 8 — бутилпропионат; 9 — ундекан (внутренний эталон)
Черт. 3
Черт. 4
(Измененная редакция, Изм. № 1, 3),
5.4. Обработку результатов анализа проводят в соответствии с разд. 4. При использовании детектора по теплопроводности относительные поправочные коэффициенты вычисляют только для этило
ввод пробы
вого и бутилового спирта, для остальных компонентов они равны единице. При использовании детектора ионизации в пламени относительные поправочные коэффициенты вычисляют для всех компонентов пробы. Массовую долю основного вещества определяют, вычитая из 100 % сумму примесей в процентах.
За результат анализа принимают среднее арифметическое двух параллельных определений. Допускаемые абсолютные расхождения между параллельными определениями при доверительной вероятности Р= 0,95 и допускаемые абсолютные расхождения между среднеарифметическими значениями параллельных определений, проведенных в разных лабораториях, указаны в табл. 1.
I
а
ев
X
а
ев
А
Б
Ж
о
V
ЗЕ
X
о.
а
о
ч
о
и
1-2
2-8
Допускаемые абсолютные расхождения между параллельными определениями, %
Этилацетат
о
ЗЕ
X
а с #
1“
ЗЕ и
0,1
0,6
X -
Б О
ас й х
о с Ч о
0,1
0,5
Бутилацетат
V
> ЗЕ
и.
Ч ЗЕ о
0,2
0,6
0,1
0,5
Таблица 1
Допускаемые абсолютные расхождения между среднеарифметическими значениями параллельных определений, проведенных в разных лабораториях, %
Этил ацетат
I о.*
о X о а ч о
и 2 и
5 зе зе
0,4
1,1
| 2 « S £
04 ВД 3S
5 о к ч ч о
0,2
0,9
Бутилацетат
OS ч и Р 2 и
§ ЗЕ ЗЕ
о7з
м
I I
К А X
go с ч ч о
0,2
0,9
^Включая этиловый и бутиловый спирт.
(Измененная редакция, Изм. № 1,3).
5.5. Определение массовой доли воды
5.5.1. Сущность метода заключается в газохроматографическом определении воды в колонке с полимерным сорбентом — полисо-рбом-1, и расчете ее массовой доли методом абсолютной градуировки.
5.5.2. Хроматограммы снимают при следующем режиме:
Для бутил ацетата Для этилацетата
Длина колонки из стекла или нержавеющей стали, м 1—2 1—2
Внутренний диаметр колонки, мм 3 3
Для бутилацетата | Для этилацетата | |
Температура термостата колонки, *С | 170-180 | 100-130 |
Температура испарителя, ®С | 180-200 | 150-170 |
Температура детектора, “С | 100 | 100 |
Расход гелия, см3/мин | 20-90 | 20-90 |
Объем пробы, вводимой в хроматограф, мм3 | 5-10 | 5-10 |
Продолжительность одного определения, мин | 7-10 | 7-10 |
Типовая хроматограмма технического этилацетата в колонке с полисорбом-1
Типовая хроматограмма технического бутил ацетата в колонке с полисорбом-1
4
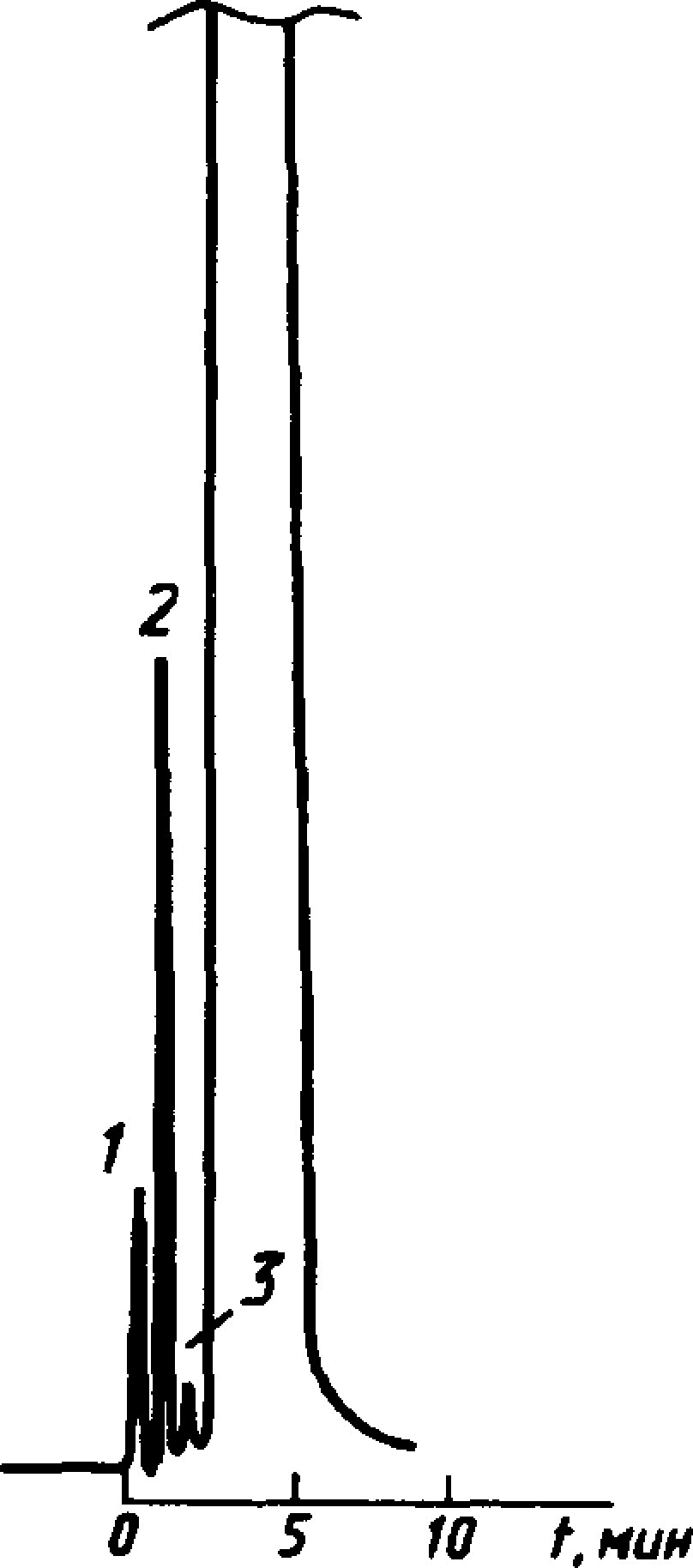
4
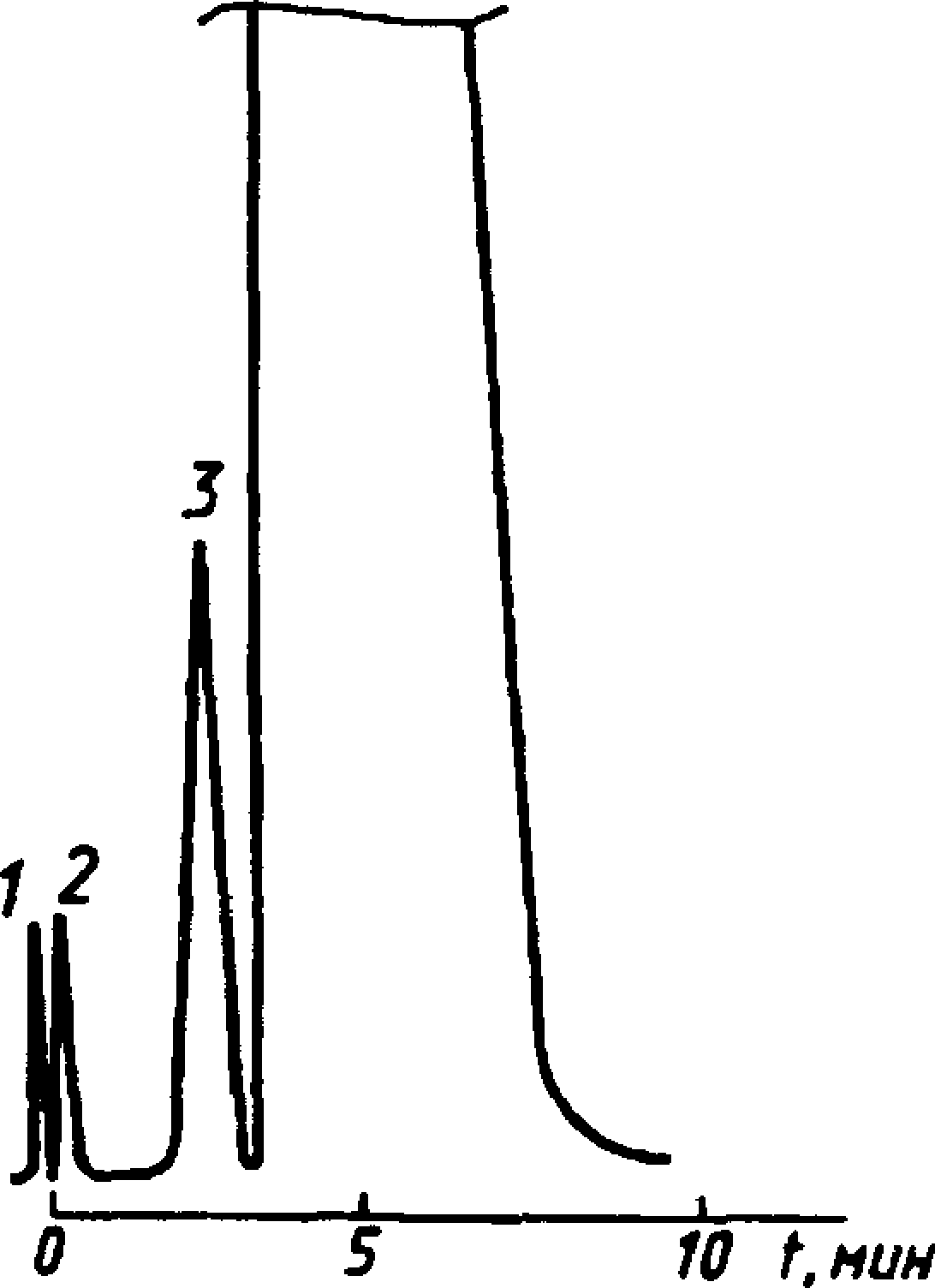
1 — воздух; 2 — вода; 3— 1 — воздух; 2 — вода; 3 — бутило-
этиловый спирт; 4 — этил- вый спирт; 4 — бутилацетат
ацетат
Черт, ja
Черт.4а
Допускается использовать другой газ-носитель, обеспечивающий определение массовой доли воды не менее 0,01 %.
Микрошприцем отбирают 5—10 мм3 пробы (объем постоянный) и снимают две хроматограммы.
Типовые хроматограммы технического этилацетата и бутилацетата в колонке с полисорбом-1 показаны на черт. За и 4а.
5.5.3. Обработку результатов анализа проводят в соответствии с п. 4.2а
За результат анализа принимают среднее арифметическое результатов двух параллельных определений, допустимые абсолютные расхождения между которыми при доверительной вероятности /*=0,95 не должны превышать 0,01 %.
5.5—5.5.3. (Введены дополнительно, Изм. № 3).
6. ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
СОСТАВА СКИПИДАРА
6.1. Сущность метода заключается в газохроматографическом разделении терпеновых углеводородов в насадочной колонке и расчете массовой доли компонентов методом внутренней нормализации.
(Измененная редакция, Изм. № 1).
6.2. Перед нанесением неподвижной жидкой фазы на твердый носитель с помощью индикаторной бумаги проверяют pH водной вытяжки трикрезилфосфата. Если pH ниже 7, трикрезилфосфат растворяют в хлороформе, нейтрализуют его раствором углекислого натрия, промывают дистиллированной водой до pH 7 и хлороформ отгоняют.
Нейтрализованный трикрезилфосфат наносят на твердый носитель в виде раствора в ацетоне.
6.3. Хроматограммы снимают при следующем режиме:
количество трикрезилфосфата от массы твердого
носителя, %................................ 20
длина колонки, м.............................. 3
внутренний диаметр колонки, мм................. 3—4
температура термостата колонки, вС................ 106
температура испарителя, “С...................... 150—200
расход гелия или азота, см3/мин.................. 40—80
объем пробы, вводимой в хроматограф, мм3............. 0,5—4
чувствительность потенциометра при использовании
детектора по теплопроводности, мВ, не более...... 5
продолжительность одного определения, мин........ 25—35
Типовые хроматограммы живичного, сульфатного, экстракционного и сухоперегонного скипидаров показаны на черт. 5—7.
Типовая хроматограмма живичного Типовая хроматограмма
скипидара (масла терпентинного) экстракционного скипидара
и очищенного сульфатного скипидара
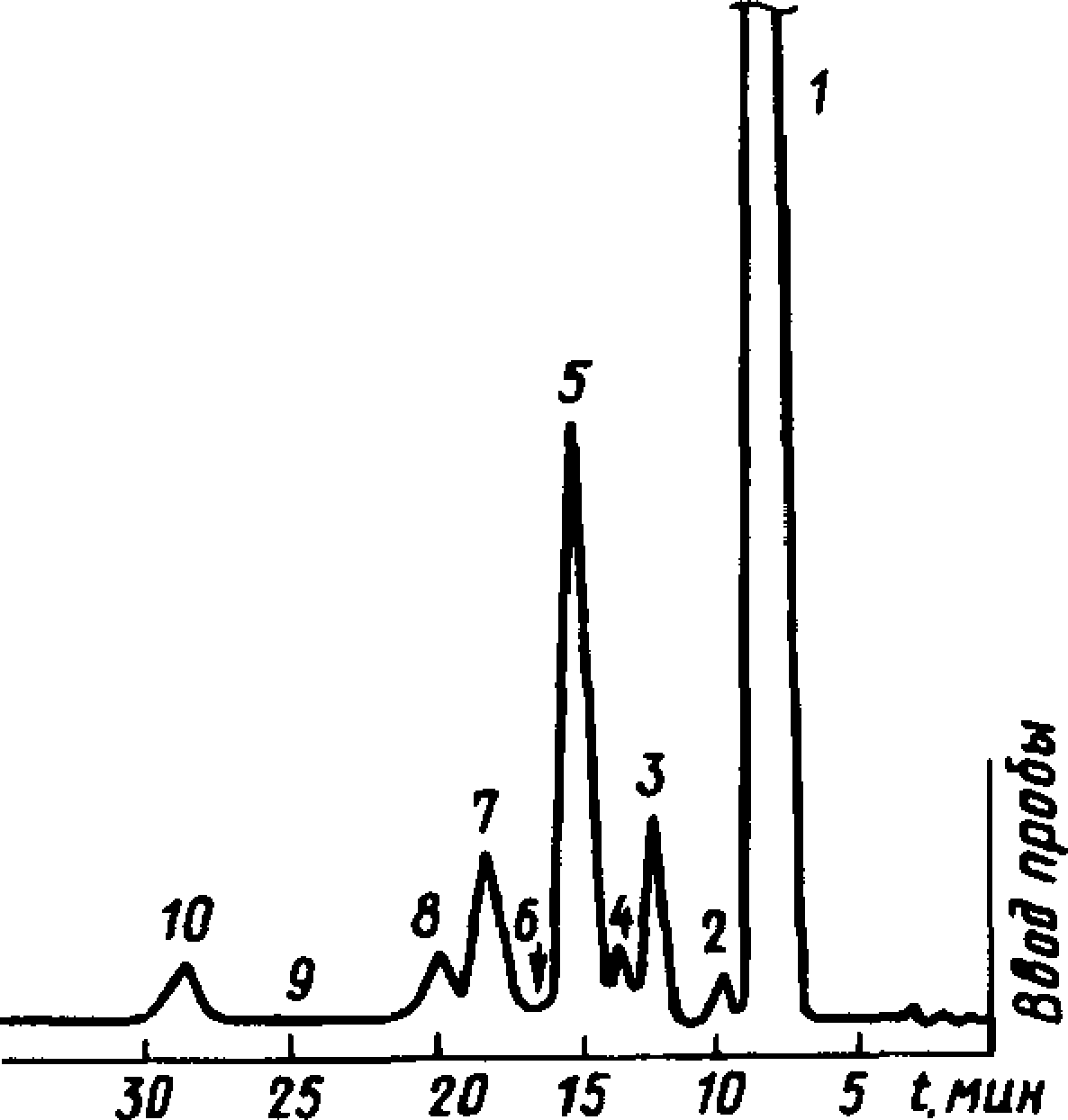
1
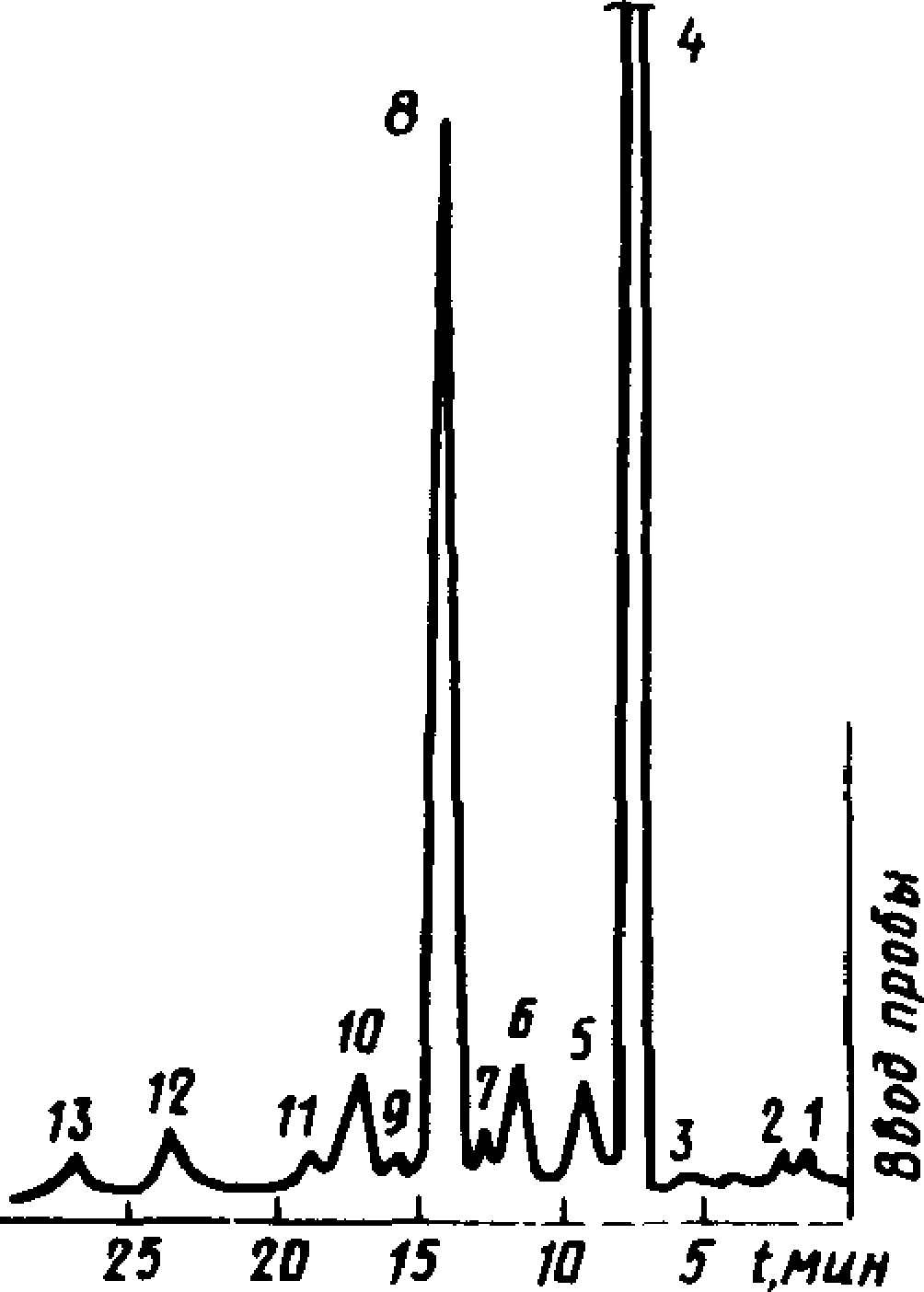
1 — а-пинен; 2—камфен; 3 — р-пинен; 4— р-мирцен; 5 — Д3-карен; 6 — а-терпинен; 7— дипентен; 8 — р-фел-ландрен; 9— л-цимол+ у-терпинен; 10 — терпинолен
Черт. 5
1,2,3— компоненты бензина; 4 — а-пинен; 5—камфен; 6— р-пинен; 7— р-мирцен; 8 — д3-карен; 9 — а-терпинен; 10— дипентен; 11 — р-фелландрен; 12 — л-цимал+ у-терпинен; 13 — терпинолен
Черт. 6
Типовая хроматограмма сухоперегонного скипидара
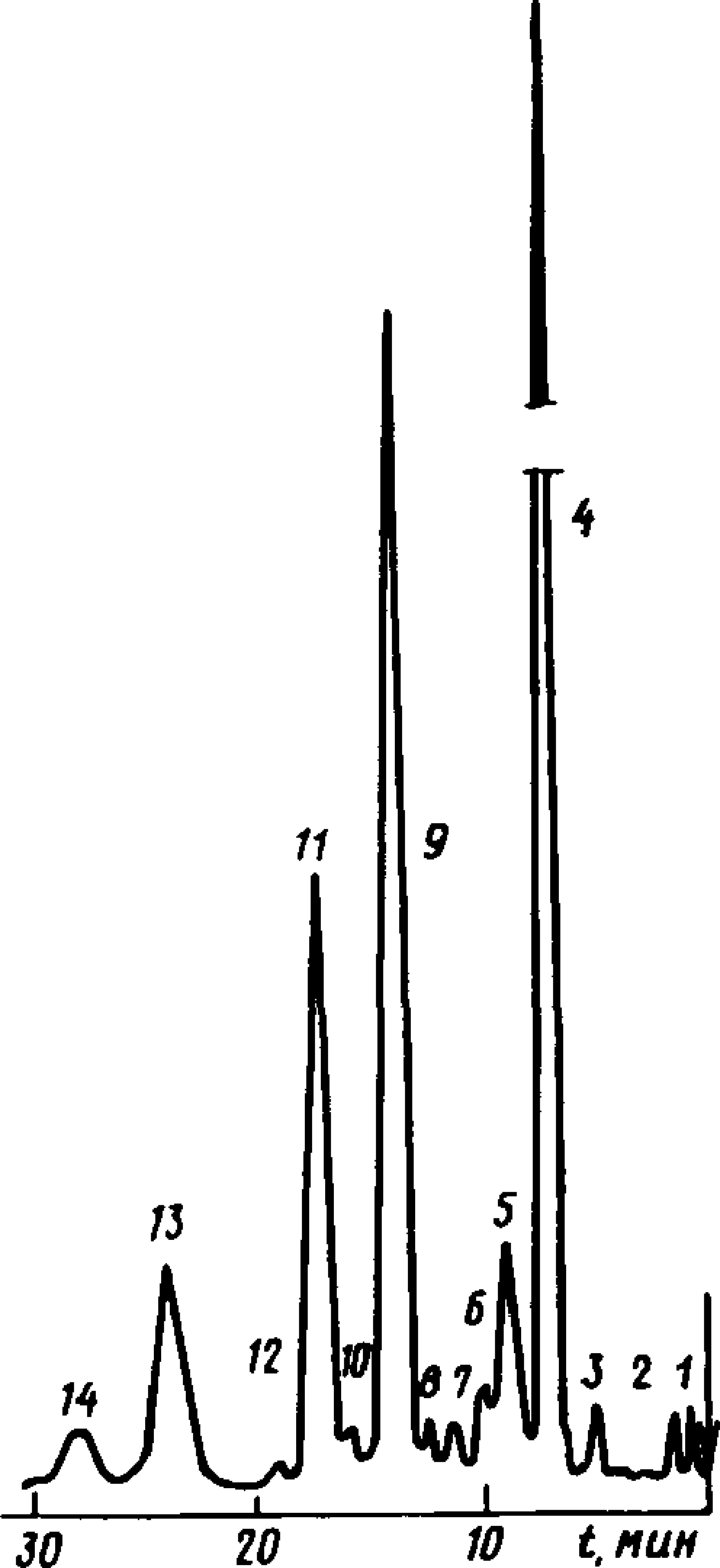
1, 2, 3, 6— неидентифицированные компоненты; 4 — а-пинен; 5 — камфен; 7 — р-пинен; 8 — р-мир-цен; 9 — Д3-карен; 10— а-терпи-нен; 11 — дипентен; 12— р-фел-ландрен; 13 — л-цимол + у-терпи-нен; 14 — терпинолен
Черт. 7
(Измененная редакция, Изм. № 1, 3).
6.4. Обработку результатов анализа проводят в соответствии с разд. 4 без введения поправочных коэффициентов, которые равны единице.
За результат анализа принимают среднее арифметическое двух параллельных определений. Допускаемые абсолютные расхождения
между параллельными определениями при доверительной вероятности Р = 0,95, а также допускаемые абсолютные расхождения между среднеарифметическими значениями параллельных определений, проверенных в разных лабораториях, указаны в табл. 2.
Таблица 2
Массовая доля компонентов, % | Допускаемые абсолютные расхождения между параллельными определениями, % | Допускаемые абсолютные расхождения между среднеарифметическими значениями параллельных определений, проверенных в разных лабораториях, % |
От 1 до 3 | 0,6 | 0,7 |
Св. 3 » 10 | 1,0 | 1,2 |
» 10 »> 40 | 1,3 | 1,5 |
» 40 » 80 | 1,8 | 2,1 |
Для определения массовой доли а- и р-пинена, полученный при расчете хроматограмм результат умножают на коэффициент К, учитывающий массовую долю нелетучего остатка.
Коэффициент К вычисляют по формуле
~ 100 -X
100 ’
где X — массовая доля нелетучего остатка, %.
При отсутствии данных о массовой доле нелетучего остатка коэффициент К для живичного и сухоперегонного скипидара принимают равным 0,98; для очищенного сульфатного скипидара — 0,99; для экстракционного скипидара — 0,965.
(Измененная редакция, Изм. № 1,2, 3).
7. ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ОСНОВНОГО ВЕЩЕСТВА И ПРИМЕСЕЙ В ТЕХНИЧЕСКОМ
КАМФЕНЕ
7.1. Сущность метода заключается в газохроматографическом разделении терпеновых углеводородов в насадочной колонке и расчете содержания компонентов методом внутренней нормализации.
7.2. Подготовку неподвижной жидкой фазы проводят по п. 6.2.
7.3. Хроматограммы снимают при следующем режиме:
количество трикрезилфосфата от массы твердого
носителя, %............................... 20
длина колонки, м.............................. 3
внутренний диаметр колонки, мм................. 3—4
температура термостата колонки, °С................ 84
температура испарителя, °С...................... 150—200
расход гелия или азота, см3/мин.................. 40—80
объем пробы, вводимой в хроматограф, мм3......... 1—5
чувствительность потенциометра при использовании
детектора по теплопроводности, мВ, не более..... 5
продолжительность одного определения, мин........ 25—35.
(Измененная редакция, Изм. № 1, 3).
7.4. Пробу камфена вводят в хроматограф в виде раствора в ацетоне с массовой долей 50 %. Типовая хроматограмма технического камфена показана на черт. 8.
(Измененная редакция, Изм. № 1, 3).
Типовая хроматограмма технического камфена
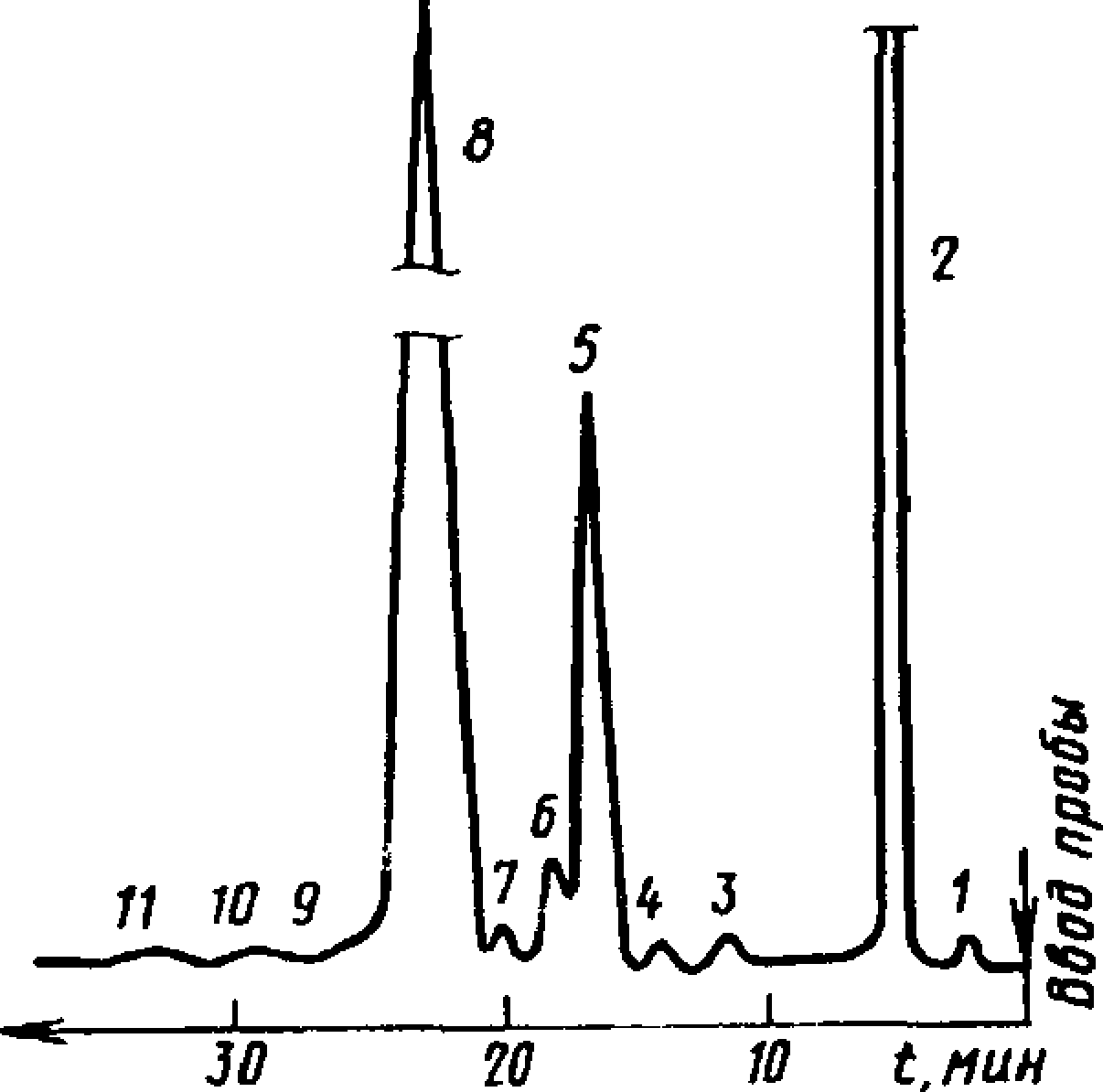
1 — воздуха; 2 — ацетон; 3 — циклофен хен; 4 — е-фенхен + у-фен-
хен+борнилен; 5 — трициклен; 6— а-пинен; 7 — а-фенхен+ р-фенхен; 8 — камфен; 9 — р-пинен+неидентифици-рованный компонент; 10— р-мирцен;
77— Д3-карен
Черт. 8
7.5. Обработку результатов анализа проводят в соответствии с разд. 4 без введения относительных поправочных коэффициентов, равных единице. За результаты анализа принимают среднее арифметическое двух параллельных определений, допускаемые абсолютные расхождения между которыми при доверительной вероятности Р = 0,95 не должны превышать: для суммы камфена и трициклена — 2,0 %, для суммы примесей — 1,0 %.
Допускаемые абсолютные расхождения между среднеарифметическими значениями параллельных определений, проведенных в разных лабораториях, не должны превышать: для суммы камфена и трициклена — 2,0 %, для суммы примесей — 1,7 %.
(Измененная редакция, Изм.
№ 1, 2, 3).
8. ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ОСНОВНОГО ВЕЩЕСТВА И ПРИМЕСЕЙ В ТЕХНИЧЕСКОЙ
КАМФАРЕ
8.1. Сущность метода заключается в газохроматографическом разделении продукта в насадочной колонке и расчете содержания компонентов методом внутренней нормализации.
8.2. Смесь полиэтиленгликоля 4000 с апиезоном L или апиезоном N наносят на твердый носитель в виде раствора в хлороформе.
8.3. Хроматограммы снимают при следующем режиме:
количество смеси полиэтиленгликоля 4000 с апиезоном L или апиезоном N от массы твердого носителя, %................................... 25
длина колонки, м.............................. 2—3
внутренний диаметр, мм........................ 3—4
температура термостата колонки, °С................ 130
температура испарителя, "С...................... 200—250
расход гелия или азота, см3/мин.................. 40—80
чувствительность потенциометра при использовании
детектора по теплопроводности, мВ, не более..... 5
объем пробы, вводимой в хроматограф, мм3......... 2—10
продолжительность одного определения, мин........ 25—35.
(Измененная редакция, Изм. № 1, 3).
8.4. Пробу камфары вводят в хроматограф в виде раствора в ацетоне с массовой долей 50 %. Типовая хроматограмма технической камфары показана на черт. 9.
(Измененная редакция, Изм. № 3, 4).
8.5. Обработку результатов анализа проводят в соответствии с разд. 4 без введения относительных поправочных коэффициентов, равных единице, вычитая из 100 % содержание воды в анализируемом продукте, определенное по ГОСТ 16399 (в качестве растворителя применяют толуол по ГОСТ 5789, толуол каменноугольный или сланцевый по ГОСТ 9880 или ксилол каменноугольный по ГОСТ 9949), в процентах. За результаты анализа принимают среднее арифметическое двух параллельных определений, допускаемые абсолютные расхождения между которыми при доверительной вероятности Р— 0,95 не должны превышать:
для камфары — 1,3 %,
для примесей с концентрацией 0,2—0,9 % — 0,2 %,
для примесей с концентрацией 1—5 % — 0,5 %.
Типовая хроматограмма технической камфары
1
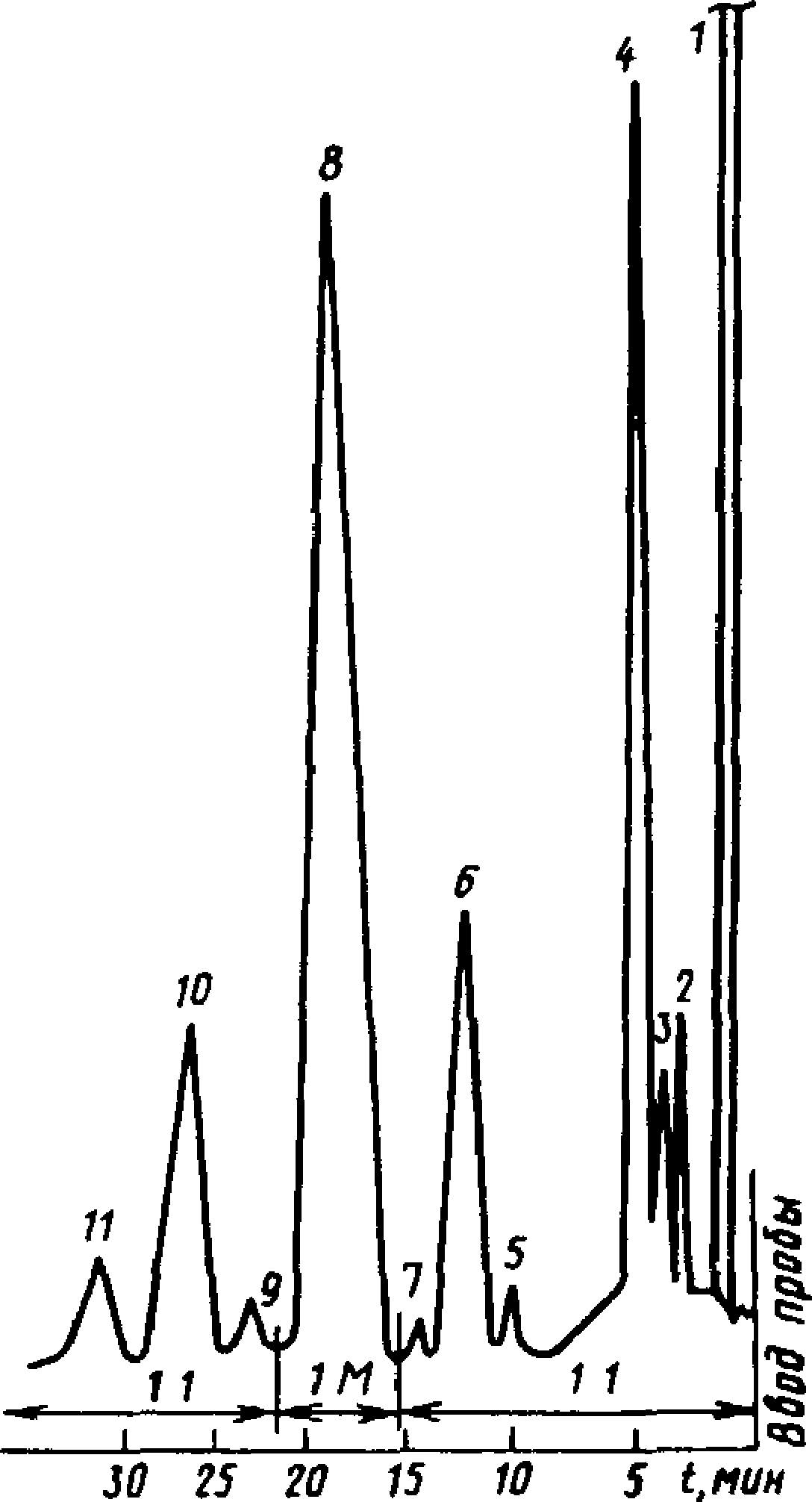
1 — ацетон, 2 — трициклен, 3 — камфен,
4 — изокамфан, 5 — фенхон, 6 — иэофен-хон, 7 и 9 — неидентифицированные компоненты, 8 — камфара, изокам
фан-5-он + и зоборнеол, 11— борнеол
Черт 9
Допускаемые абсолютные расхождения между среднеарифметическими значениями параллельных определений, проведенных в разных лабораториях, не должны превышать: для камфары — 1,3 %,
для примесей с концентрацией 0,2—0,9 % — 0,2 %, для примесей с концентрацией 1—5 % — 0,7 %.
(Измененная редакция, Изм. № 2, 3).
9. ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ СМОЛЯНЫХ КИСЛОТ В КАНИФОЛИ
9.1. Метод заключается в газохроматографическом определении массовой доли смоляных кислот в различных видах канифоли методом внутреннего стандарта. Смоляные кислоты анализируют в виде метиловых эфиров, полученных при термораспаде их тетраметилам-мониевых солей в испарителе хроматографа при температуре 280— 300 “С. Вещества нейтрального характера, мешающие определению смоляных кислот, предварительно экстрагируют бензолом из водноспиртового раствора солей тетраметиламмония.
9.2. Подготовка к анализу
9.2.1. Приготовление насадки и заполнение колонки проводят в соответствии с пп. 2.1 и 2.3.
9.2.2. Прибор градуируют по градуировочным смесям абиетиновой кислоты и кислоты стандарта (маргариновой или стеариновой). Градуировочные коэффициенты остальных смоляных кислот принимают равными градуировочному коэффициенту абиетиновой кислоты.
9.2.2.1. Получение абиетиновой кислоты
Абиетиновую кислоту (массовая доля основного вещества не менее 90 %) выделяют из живичной канифоли путем пятикратной перекристаллизации из этилового спирта.
В стеклянном стакане вместимостью 250 см3 взвешивают (30+5) г живичной канифоли, растворяют в 60—80, см3 этилового спирта, добавляют 2—3 капли концентрированной соляной кислоты и кипятят в открытом стакане на водяной бане в течение 1 ч. Выпавший после охлаждения при комнатной температуре осадок абиетиновой кислоты переносят на воронку Бюхнера и с помощью водоструйного насоса отсасывают маточный раствор. Перекристаллизацию проводят еще четыре раза. Высушенный на фильтре осадок используют для определения градуировочных коэффициентов. Хранить абиетиновую кислоту следует в запаянной ампуле в защищенном от света месте.
9.2.2.2. Определение массовой доли абиетиновой кислоты
Массовую долю абиетиновой кислоты в выделенном препарате
определяют методом внутренней нормализации.
(0,20±0,05) г абиетиновой кислоты взвешивают, растворяют в 1 см3 этилового спирта, добавляют одну каплю фенолфталеина и титруют из бюретки раствором гидроокиси тетраметиламмония до появления малиновой окраски. Полученный раствор упаривают на
водяной бане до желеобразного состояния, добавляют несколько капель этилового спирта и с помощью микрошприца вводят в хроматограф. Хроматограммы (не менее трех) снимают в условиях, приведенных в п. 9.2.2.3.
Массовую долю абиетиновой кислоты рассчитывают по формуле, приведенной в п. 4.2.
Массовую долю основного вещества в кислоте-стандарте определяют по формуле, приведенной в п. 4.2.
9.2.2.3. Режим работы хроматографа при градуировке и проведении испытаний
Массовая доля неподвижной жидкой фазы ПДЭГС, %;
для носителей типа хроматон N-Aco-DMCS.......... 6
для носителей типа инертон Аю-DMCS............. 3
Длина колонки, м............................. 3
Внутренний диаметр колонки, мм................. 3
Температура термостата, °С...................... 190—195
Температура испарителя, °С...................... 280—300
Объемный расход гелия, см3/мин.................. 30—80
Объемный расход водорода, см3/мин............... 30—60
Объемный расход воздуха, см3/мин................ 300—350
Объем пробы при градуировке и анализе, мм3........ 0,5—2,0
Масса анализируемой пробы, г................... 0,20—0,25
Массовая доля кислоты-стандарта, %............... 5—6
Продолжительность анализа, мин.................. 35—50
9.2.2.4. Градуировка прибора
Смеси абиетиновой кислоты и кислоты-стандарта готовят в следующих соотношениях 0,5:1, 1:1, 2:1, 3:1. Абиетиновую кислоту и кислоту-стандарт взвешивают в стаканчике для взвешивания, записывая результаты взвешиваний в граммах с точностью до четвертого десятичного знака.
Масса кислоты-стандарта — 0,1 г, масса абиетиновой кислоты — 0,05—0,30 г.
Смесь кислот превращают в соли тетраметиламмония в соответствии с п. 9.2.2.2.
Каждую градуировочную смесь хроматографируют не менее семи раз.
Относительный градуировочный коэффициент вычисляют по формуле, приведенной в п. 2.5.
Массу абиетиновой кислоты и кислоты-стандарта рассчитывают с учетом массовой доли основного вещества.
9.2.3. Проведение анализа
В стаканчике для взвешивания взвешивают 0,20—0,25 г канифоли и 0,010—0,015 г маргариновой или стеариновой кислоты (результаты взвешивания записывают с точностью до четвертого десятичного знака). Приготовленную смесь растворяют в 5 см3 этилового спирта, добавляют 2—3 капли фенолфталеина, титруют из бюретки водным раствором гидроокиси тетраметиламмония до появления устойчивой малиновой окраски и добавляют воду в таком количестве, чтобы соотношение объемов спирт-вода составляло 1:1. Раствор переносят в делительную воронку, добавляют 10 см3 бензола, экстрагируют нейтральные вещества и после расслаивания 1—2 см3 нижнего водноспиртового слоя переносят пипеткой в стаканчик для взвешивания и помещают в водяную баню.
Раствор солей упаривают до желеобразного состояния, добавляют этиловый спирт в таком объеме, чтобы раствор можно было набрать в микрошприц. Микрошприцем набирают 0,5—2,0 мм3 раствора солей тетраметиламмония и вводят в испаритель хроматографа. Хроматограммы снимают при условиях, изложенных в п. 9.2.2.3.
На черт. 10, 11, 12 представлены типичные хроматограммы живичной, экстракционной и талловой канифоли.
9.2.4. Обработка результатов
Массовую долю смоляных кислот в канифоли вычисляют по формуле, приведенной в п. 4.1.
За результат анализа принимают среднее арифметическое результатов двух параллельных определений, допускаемые абсолютные расхождения между которыми при доверительной вероятности Р = 0,95 приведены в табл. 3.
Таблица 3
Массовая доля смоляной кислоты, % | Допустимые расхождения результатов двух параллельных определений при внутрила-бораторном контроле, % |
0,1-0,7 | 0,15 |
0,8-4,0 | 0,4 |
5-10 | 0,7 |
11-20 | 1,2 |
21-40 | 2,5 |
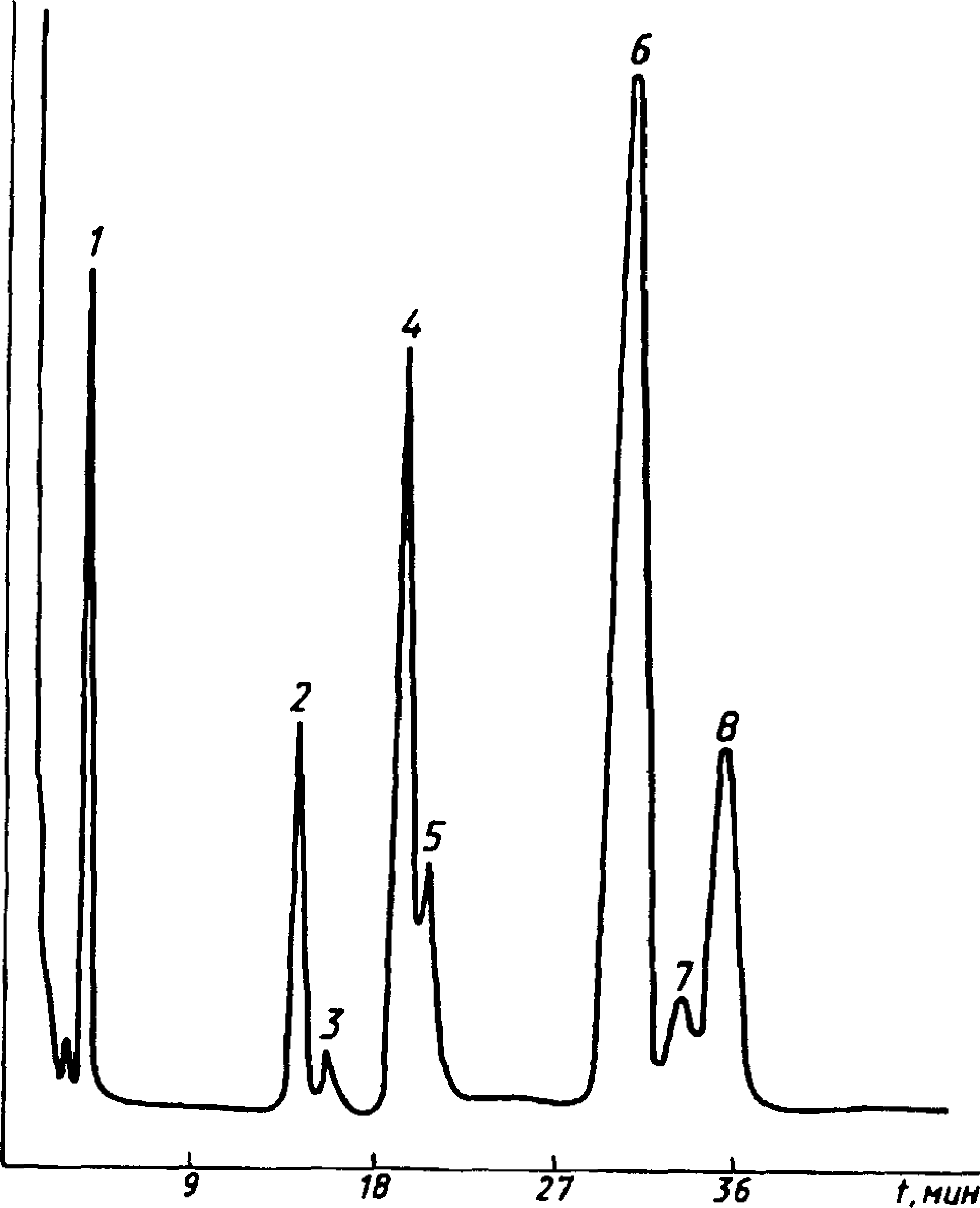
/ — метилмаргарат; 2 — метилпимарат; 3 — метилсандарако-пимарат; 4 — метилпалюстрат; 5 — метилизопимарат; 6 — ме-тилабиетат; 7 — метилдегидроабиетат; 8 — метилнеоабиетат
Черт. 10
1 — метилмарганат; 2 — метил пи Марат; 3 — метилсандарако-п и Марат; 4 — метилпалюстрат; 5 — метил изопимарат; 6 — ме-тилабиетат; 7 — метилдегидроабиетат; 8 — метилиеоабиетат
Черт. 11
1 — метилмаргарат; 2— метилпимарат; 3 — метилсандаракопимарат; 4 — метил пал юстрат; 5 — метил изопимарат; 6 — метилабиетат;
7 — метилдегидроабиетат; 8 — метилнеоабиетат
Черт. 12
Разд. 9. (Введен дополнительно, Изм. № 4).
ИНФОРМАЦИОННЫЕ ДАННЫЕ
1. РАЗРАБОТАН И ВНЕСЕН Министерством лесной, целлюлозно-бумажной и деревообрабатывающей промышленности СССР
РАЗРАБОТЧИКИ
А.М. Чащин, Л.В. Косюкова
2. УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Государственного комитета стандартов Совета Министров СССР от 03.02.76 № 299
3. Срок проверки — 1996 г. Периодичность проверки — 5 лет.
4. ВВЕДЕН ВПЕРВЫЕ
5. ССЫЛОЧНЫЕ НОРМАТИВНО-ТЕХНИЧЕСКИЕ ДОКУМЕНТЫ
Обозначение НТД, на который дана ссылка | Номер пункта, подпункта |
1.1 | |
1.1 | |
1Л | |
1Л | |
1Л | |
1Л | |
1Л | |
1.1, 8.5 | |
1Л | |
1.1 | |
1.1 | |
1.1 | |
1.1 | |
1.1 | |
1.1 | |
1.1 | |
8.5 | |
8.5 | |
8.5 | |
1.1 |
Продолжение
Обозначение НТД, на который дана ссылка | Номер пункта, подпункта |
1.1 | |
1.1 | |
1.1 |
6. Ограничение срока действия снято Постановлением Госстандарта СССР от 09 08.91 № 1338
7. ПЕРЕИЗДАНИЕ (июнь 1997 г.) с Изменениями № 1, 2, 3, 4, утвержденными в ноябре 1981 г., апреле 1986 г., августе 1991 г., феврале 1993 г. (ИУС 2-82, 7-86, 11-91, 8-93)
Редактор В.Н. Копысов Технический редактор Н.С. Гришанова Корректор М.С. Кабашова Компьютерная верстка В.И. Грищенко
Изд. лиц. №021007 от 10.08.95. Сдано в набор 24.07.97. Подписано в печать 03.09.97. Уел. печ. л. 1,63. Уч.-изд. л. 1,45. Тираж 128 экз. С851. Зак. 624.
ИПК Издательство стандартов, 107076, Москва, Колодезный пер., 14. Набрано в Издательстве на ПЭВМ
Филиал ИПК Издательство стандартов — тип. "Московский печатник"
Москва, Лялин пер., 6.
Плр № 080102
