
ГОСТ Р 52896-2017
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРОИЗВОДСТВО ЛЕКАРСТВЕННЫХ СРЕДСТВ
Производственные системы и оборудование для производства лекарственных средств. Общие требования
Manufacturing of medicinal product. Manufacturing systems and equipment for the production of medicines. General requirements
ОКС 13.060.70
Дата введения 2018-07-01
Предисловие
1 РАЗРАБОТАН Государственным бюджетным образовательным учреждением высшего профессионального образования Первым Московским государственным медицинским университетом имени И.М.Сеченова Министерства здравоохранения Российской Федерации (Первый МГМУ имени И.М.Сеченова)
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 458 "Разработка, производство и контроль качества лекарственных средств"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 8 сентября 2017 г. N 1036-ст
4 Настоящий стандарт разработан с учетом основных нормативных положений стандарта АСТМ Е2500-13:2013* "Стандартное руководство по разработке спецификаций, проектированию и верификации фармацевтических и биофармацевтических производственных систем и оборудования" (ASTM Е2500-13:2013 "Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment", NEQ)
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
5 ВЗАМЕН ГОСТ P 52896-2007
6 ПЕРЕИЗДАНИЕ. Июнь 2019 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Настоящий стандарт описывает научно обоснованный подход, основанный на оценке риска, к разработке спецификаций, проектированию и верификации производственных систем и оборудования, которые способны оказывать влияние на качество продукции и безопасность пациентов.
Настоящий стандарт описывает системный, эффективный и рациональный подход к обеспечению пригодности производственных систем и оборудования для предполагаемого использования, а также обеспечению того, что риски, связанные с качеством продукции и, соответственно, с безопасностью пациентов, эффективно управляются в той степени, в которой на них оказывают влияние упомянутые системы и оборудование.
Основной целью работ по созданию производственных систем и оборудования является создание таких систем, которые в состоянии поддерживать определенные и контролируемые технологические (и другие) процессы, обеспечивающие стабильное производство продукции, соответствующей установленным требованиям к ее качеству.
Подход, описанный в настоящем стандарте, также поддерживает непрерывное улучшение возможностей процессов и способствует внедрению инновационных решений, например процессно-аналитической технологии (далее - ПАТ).
Стандарт включает в себя описание:
- основных концепций, которые следует применять при выполнении работ по созданию производственных системы и оборудования для фармацевтической промышленности,
- процесса разработки спецификаций, проектирования и верификации;
- необходимых вспомогательных процессов.
Описанный в настоящем стандарте подход направлен на выполнение международных регуляторных ожиданий по обеспечению пригодности производственных систем и оборудования для предполагаемого назначения и удовлетворения требований, предъявляемых к проектированию, монтажу, эксплуатации и эксплуатационным характеристикам.
Настоящий стандарт согласован собщими принципами, описанными в руководствах [1 ], [2], [3] и [4].
1 Область применения
Настоящий стандарт применим ко всем элементам фармацевтических и биофармацевтических производственных систем, в том числе к инженерно-техническому оборудованию, производственному оборудованию, вспомогательным системам, связанным с ними системам мониторинга и контроля, а также системам автоматизации, которые способны оказывать влияние на качество продукции и безопасность пациента (далее - производственные системы).
Также данный стандарт может быть применим к лабораторным и информационным производственным системам, к производственным системам медицинского оборудования.
Настоящий стандарт применим как к новым, так и к уже существующим производственным системам. Описанный в стандарте подход может использоваться для внедрения изменений в существующие системы и их непрерывного улучшения в процессе эксплуатации.
Настоящий стандарт применим к производственным системам на протяжении всего их жизненного цикла, от разработки концепции до прекращения эксплуатации.
2 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
2.1 критерии приемлемости: Критерии, которым должна соответствовать система или ее компонент, для того чтобы быть принятой пользователем или иным уполномоченным лицом.
2.2 анализ проекта: Запланированный и систематический пересмотр спецификаций, проекта и проектных решений, а также постоянное внедрение улучшений, осуществляемое, при необходимости, в течение всего жизненного цикла производственной системы. В рамках анализа проекта осуществляются оценка результата работы на соответствие стандартам и требованиям, выявление проблем и предложение необходимых корректирующих действий.
2.3 производственные системы: Элементы фармацевтических и биофармацевтических производственных мощностей, включая производственные системы, инженерно-техническое оборудование, технологическое оборудование, вспомогательные системы, связанные с ними системы мониторинга и контроля, а также системы автоматизации, которые способны оказывать влияние на качество продукции и безопасность пациента.
2.4 специалисты в конкретной области (subject matter experts, SMEs): Лица, обладающие специальными знаниями и отвечающие за конкретную область или направление работы (например, служба качества, инженерные системы, автоматизация, разработка, эксплуатация и так далее).
2.5 верификация: Системный подход к проверке того факта, что производственные системы (как по отдельности, так и в совокупности) пригодны для предполагаемого использования, смонтированы и функционируют надлежащим образом. Данный термин обобщает все подходы к оценке пригодности систем к использованию по предполагаемому назначению (например, квалификация, приемка и ввод в эксплуатацию, верификация, валидация системы и пр.).
3 Общие требования
В настоящем стандарте используются следующие ключевые концепции:
- подход, основанный на оценке риска;
- научно обоснованный подход;
- критические аспекты производственных систем;
- программируемое качество;
- надлежащая инженерная практика;
- специалист в конкретной области;
- использование документации производителя;
- непрерывное улучшение процесса.
3.1 Подход, основанный на оценке риска
3.1.1 Управление риском должно лежать в основе и надлежащим образом использоваться на каждом этапе процесса разработки спецификаций, проектирования и верификации.
3.1.2 Два основных принципа управления риском при обеспечении качества приведены в руководстве [2].
3.1.2.1 Оценка риска для качества продукции должна основываться на научных знаниях и, в конечном счете, быть связана с защитой пациента.
3.1.2.2 Степень усилий, соблюдения надлежащих процедур и документирования процесса управления риском при обеспечении качества должны быть соизмеримы с уровнем риска.
3.1.3 Вышеприведенные принципы должны применяться по отношению к разработке спецификаций, проектированию и верификации производственных систем.
3.1.4 Диапазон и степень управления риском при обеспечении качества на этапах разработки спецификаций, проектирования и верификации, а также степень документирования процесса должны основываться на риске для качества продукции и безопасности пациента.
3.2 Научно обоснованный подход
3.2.1 Информация о продукции и процессе вследствие их связи с качеством продукции и безопасностью пациента должна использоваться в качестве основы для принятия научно обоснованных решений, основанных на оценке риска, которые обеспечивают пригодность производственных систем для предполагаемого использования (на этапе проектирования и последующей верификации).
3.2.2 Примеры подобной информации о продукции и процессе включают в себя: критические показатели качества (critical quality attributes, CQAs), критические параметры процесса (critical process parameters, CPPs), информацию о стратегии системы управления процессом и имеющийся опыт производства.
3.3 Критические аспекты производственных систем
3.3.1 Критическими аспектами производственных систем обычно являются функции, свойства, возможности и характеристики, необходимые производственному процессу и системам для обеспечения стабильного качества продукции и безопасности пациента. Данные аспекты должны быть выявлены и задокументированы основываясь на научном понимании продукции и процесса.
3.3.2 В дальнейшем в тексте настоящего руководства вышеупомянутые элементы для краткости будут обозначаться как критические аспекты.
3.3.3 Деятельность по верификации должна быть сосредоточена на данных аспектах производственных систем и должна быть задокументирована. Процесс верификации определен в пункте 7.4.
3.4 Программируемое качество
3.4.1 Для обеспечения включения критических аспектов в системы на этапах разработки спецификаций и проектирования следует использовать концепции "программируемого качества". Критические аспекты проекта и связанные с ними критерии приемлемости должны быть задокументированы.
3.4.2 Уверенность в том, что производственные системы пригодны для предполагаемого использования не должны основываться исключительно на верификации после окончания монтажа, но также достигаться путем спланированного и структурированного подхода к верификации, применяемого на протяжении всего жизненного цикла системы.
3.5 Надлежащая инженерная практика
3.5.1 В основе процесса разработки спецификаций, проектирования и верификации должна лежать надлежащая инженерная практика (Good Engineering Practice, GEP).
3.5.2 Надлежащая инженерная практика - определенные инженерные методы и стандарты, которые применяются на протяжении всего жизненного цикла производственной системы для выработки приемлемых и эффективных решений.
3.5.3 Примеры надлежащей инженерной практики включают в себя:
3.5.3.1 При осуществлении деятельности по разработке спецификаций, проектированию и монтажу должны приниматься во внимание все релевантные требования, в том числе надлежащих практик (GxP), безопасности, охраны здоровья и окружающей среды, эргономики, эксплуатации, обслуживания, признанные отраслевые стандарты и прочие нормативные требования.
3.5.3.2 В спецификации, проекты, документы материально-технического снабжения и прочую контрактную документацию необходимо включать соответствующие положения о качестве.
3.5.3.3 Необходимо сформировать документацию, включающую в себя весь жизненный цикл производственной системы, в том числе планирование, разработку спецификаций, проектирование, верификацию, монтаж, приемку и обслуживание.
3.5.3.4 Надлежащая степень надзора и контроля должна быть достигнута путем адекватной верификации процессов выполнения, сооружения и монтажа.
3.6. Специалисты в конкретной области
3.6.1 Специалисты в конкретной области - это лица, обладающие специальными знаниями и отвечающие за конкретную область или направление работы (например, служба качества, организация производства, автоматизация, разработка, эксплуатация и так далее).
3.6.2 Специалисты в конкретной области, в соответствии с их направлением работы и должностными обязанностями, должны принимать ведущее участие в верификации производственных систем.
3.6.3 В обязанности специалистов в конкретной области входят планирование и определение стратегий по верификации, установление критериев приемлемости, выбор надлежащих методов испытаний, выполнение верификационных испытаний и анализ результатов.
3.7 Использование документации производителя
3.7.1 Документация производителя, включая документацию по испытаниям, может быть использована в качестве части документации по верификации в том случае, если компания, осуществляющая верификацию, обратилась к производителю и получила данные о:
3.7.1.1 приемлемости системы обеспечения качества производителя,
3.7.1.2 технических возможностях производителя и
3.7.1.3 применении производителем надлежащей инженерной практики, что может служить подтверждением точности и пригодности для целей верификации полученной от производителя информации.
3.7.2 В случае выявления несоответствий в системе обеспечения качества, технических возможностях производителя или применении им ненадлежащей инженерной практики компания, осуществляющая верификацию, может принять решение о нивелировании потенциальных рисков путем осуществления конкретных, целенаправленных дополнительных верификационных проверок или применения прочих методов контроля, нежели путем повторения действий производителя и воспроизведением его документации.
3.7.3 Принятие решения об использовании документации производителя (и обоснование данного решения) с целью поддержания верификации критических аспектов компонента производства должно основываться на предполагаемом назначении производственной системы, должно быть задокументировано и одобрено специалистами в конкретной области, в том числе представителями службы качества.
3.8 Непрерывное улучшение процесса
3.8.1 Поскольку в процессе промышленного производства происходит накопление опыта, необходимо выявлять возможности для совершенствования, основываясь на периодических проверках и оценочных испытаниях, данных об эксплуатации и эксплуатационных характеристиках, а также на анализе первопричин сбоев в работе.
3.8.2 Управление изменениями должно предоставлять надежный механизм для безотлагательного внедрения технически обоснованных усовершенствований, принимая во внимание описанный в настоящем руководстве подход к разработке спецификаций, проектированию и верификации.
4 Требования к основному процессу
4.1 Общие положения
Процесс разработки спецификаций, проектирования и верификации производственных систем должен включать в себя следующие этапы:
- определение требований;
- разработка спецификаций и проектирование;
- верификация;
- приемка и допуск.
4.1.1 Положения надлежащей инженерной практики должны применяться на протяжении всего процесса.
4.1.2 Управление рисками должно проводиться таким образом, чтобы оценить риски, связанные с качеством продукции и безопасностью пациентов, возникновение которых зависит от производственной системы и соответствующего проектного решения. Управление рисками представляет собой вспомогательный процесс, который описан в пункте 8.2.
4.1.3 Анализ проекта должен повторяться на протяжении всего жизненного цикла производственной системы. Процесс анализа проекта является вспомогательным и описан в пункте 8.3.
4.1.4 Управление изменениями должно применяться на протяжении всего процесса. Управление изменениями представляет собой вспомогательный процесс, который описан в пункте 5.4.
4.2 Определение исходных требований к производственной системе
4.2.1 Необходимо выявить конкретные требования, которые лягут в основу дальнейшей разработки спецификаций, проектирования и верификации производственной системы.
4.2.2 Данные конкретные требования, относящиеся к качеству продукции и безопасности пациентов, должны быть основаны на:
4.2.2.1 научном понимании и данных о продукции,
4.2.2.2 научном понимании и данных о процессе,
4.2.2.3 регуляторных требованиях и
4.2.2.4 требованиях к уровню качества самой компании.
4.2.3 Научное понимание и данные о продукции и процессе, в том числе данные об источниках изменчивости, идентификация критических параметров качества и информация о стратегии системы управления процессом должны основываться на научных данных, полученных в ходе экспериментальной и опытно-конструкторской работы, а также на опыте производства. Данные о продукции и процессе образуют основу научного понимания, как описано в руководствах [1], [4] и в стандарте [5].
|
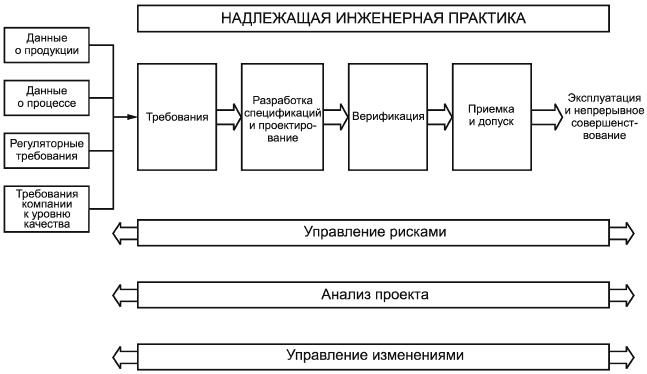
Рисунок 1 - Процесс разработки спецификаций, проектирования и верификации
4.3 Разработка спецификаций и проектирование производственной системы
4.3.1 Компании должны разработать надлежащие механизмы передачи исходных требований, в том числе критериев качества продукции, лицам, ответственным за проектирование, чтобы производственная система была спроектирована с учетом релевантных данных о продукции, процессе и прочих требованиях. Практические подходы к проектированию процессов с использованием процессно-аналитической технологии описаны в стандарте [6].
4.3.2 Деятельность по разработке спецификаций и проектированию должна включать в себя акцент на тех аспектах, которые были выявлены как критические для качества продукции и безопасности пациентов. Данные критические аспекты производственной системы должны быть установлены и задокументированы специалистами в конкретной области.
4.4 Проведение верификации
Должен быть сформулирован системный подход, позволяющий проверить тот факт, что производственные системы (как по отдельности, так и в совокупности) пригодны для предполагаемого использования, смонтированы и функционируют надлежащим образом. Данный подход должен быть сформулирован и задокументирован. Степень верификации и степень подробности документации должны основываться на рисках, связанных, помимо прочего, с качеством продукции и безопасностью пациента, а также сложности и новизне производственной системы. Информация о верификации содержится в стандартах [7] и [8], а также в руководстве FDA для производителей. Валидация процессов: основные принципы и подходы.
4.4.1 Критерии приемлемости
4.4.1.1 Критерии приемлемости представляют собой показатели, которым должна соответствовать производственная система, для того чтобы быть пригодной к предполагаемому применению и чтобы быть принятой пользователем или иным уполномоченным лицом.
4.4.1.2 Критерии приемлемости должны быть определены специалистами в конкретной области.
4.4.1.3 Критерии приемлемости для критических (с точки зрения качества продукции и безопасности пациентов) аспектов должны быть одобрены службой качества.
4.4.2 Стратегия верификации
4.4.2.1 Критерии приемлемости и стратегия верификации должны быть задокументированы в надлежащих планах по верификации.
4.4.2.2 В планах по верификации должно быть определено, что представляет собой приемлемая документация последующей деятельности по верификации.
4.4.2.3 План по верификации должен быть разработан и одобрен специалистами в конкретной области. Планы по верификации систем, включающих критические аспекты, должны быть одобрены службой качества.
4.4.3 Деятельность по верификации
4.4.3.1 Специалисты в конкретной области должны осуществлять деятельность по верификации или контролировать ее и документировать результаты верификации таким образом, как это было оговорено в планах по верификации.
4.4.3.2 При верификации может быть использована документация производителя, как описано в пункте 6.8.
4.4.3.3 Завершение деятельности по верификации должно быть задокументировано.
4.4.4 Анализ результатов
4.4.4.1 Вся готовая документация по верификации должна быть проанализирована независимым специалистом (специалистами) в конкретной области, имеющим надлежащую квалификацию, который не принимал участия в проведении верификационных испытаний.
4.4.4.2 Лица, осуществляющие анализ, должны убедиться, что все испытания были завершены и надлежащим образом задокументированы.
4.4.4.3 Отступления и отклонения от планов верификации должны быть рассмотрены и урегулированы лицом, осуществляющим анализ, и/или прочими подходящими специалистами в конкретной области.
4.5 Приемка и выпуск
4.5.1 Специалисты в конкретной области должны подтвердить, что производственная система пригодна для использования. Данное подтверждение должно быть задокументировано.
4.5.2 В документацию должны быть включены анализ или обзор результатов, и анализ любых несоответствий заданным критериям приемлемости для критических аспектов.
4.5.3 В документацию должно быть включено однозначное заявление, основывающееся на упомянутом анализе, о том, пригодна ли производственная система для предполагаемого использования. Лица, задействованные в определении данного факта, должны быть идентифицированы и внесены в документацию.
4.5.4 Подобную документацию должны подготавливать и одобрять специалисты в конкретной области. Данная документация для систем, содержащих критические аспекты, должна быть одобрена службой качества.
4.5.5 После указанных одобрений производственная система может быть допущена в эксплуатацию.
5 Вспомогательные процессы
5.1 Общие положения
Процесс разработки спецификаций, проектирования и верификации должен опираться на управление рисками, анализ проекта и управление изменениями, как описано в нижеприведенных подпунктах.
5.2 Управление риском при обеспечении качества
5.2.1 Управление риском при обеспечении качества представляет собой системный процесс, направленный на оценку, контроль, предоставление информации и анализ рисков, относящихся к качеству лекарственного средства и безопасности пациента.
5.2.2 Оценки риска должны проводиться на надлежащих этапах, чтобы определить риски в отношении качества продукции и безопасности пациента, связанные с производственными системами и соответствующими проектными решениями.
5.2.3 Риски, касающиеся доставки, в том числе риски, связанные с поставщиком или строительством, а также риски, связанные с технологической новизной или сложностью, следует рассматривать с точки зрения их наибольшего влияния на качество продукции и безопасность пациентов.
5.2.4 Оценки риска должны осуществляться подходящими специалистами в конкретной области.
5.2.5 На основе оценок риска должны быть выбраны надлежащие способы контроля и верификации, чтобы свести риск к приемлемому уровню, при этом акцент следует делать на рисках, связанных с критическими аспектами производственной системы.
5.2.6 Степени контроля и верификации должны быть соизмеримы с уровнем риска для качества продукции и безопасности пациента.
5.2.7 Необходимо предусмотреть иные пригодные механизмы контроля рисков том случае*, если их не удается избежать при проектировании.
________________
* Текст документа соответствует оригиналу. - .
5.2.8 Более подробная информация по управлению рисками приведена в руководстве [2] и стандарте [9].
5.3 Анализ проекта
5.3.1 Анализ проекта - запланированный и систематический пересмотр спецификаций, проекта и проектных решений, а также постоянное внедрение улучшений, осуществляемое, при необходимости, в течение всего жизненного цикла производственной системы.
5.3.2 В рамках анализа проекта осуществляется оценка результата работы на соответствие стандартам и требованиям, выявление проблем и предложение необходимых корректирующих действий.
5.3.3 Анализ проекта осуществляется для удостоверения в том, что:
5.3.3.1 Проект удовлетворяет требования, предъявляемые к продукции и процессу;
5.3.3.2 Должное внимание уделено критическим аспектам производственных систем;
5.3.3.3 Были выявлены риски, связанные с качеством продукции и безопасностью пациента;
5.3.3.4 Неприемлемые риски минимизированы за счет проекта или другим образом.
5.3.4 Анализ проекта должен производиться надлежащими специалистами в конкретной области.
5.3.5 Анализ проекта должен быть задокументирован. Лица, осуществляющие анализ, должны быть идентифицированы.
5.3.6 Документация по анализу проекта должна включать в себя заявление о том, что затрагиваемые элементы являются приемлемыми, при условии выполнения предлагаемых корректирующих действий.
5.4 Управление изменениями
5.4.1 Процессы управления изменениями должны быть разработаны и применяться на протяжении всего жизненного цикла.
5.4.2 До приемки следует применять управление изменениями. Данный процесс должен управляться (а изменения - быть одобрены) специалистами в конкретной области. Об изменениях, влияющих на критические аспекты производственных систем, необходимо проинформировать службу качества.
5.4.3 После приемки, но до начала промышленного производства, следует применять оперативное управление изменениями.
Библиография
[1] Руководство ICH Q8 Фармацевтическая разработка
_______________
Доступно в Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH), Секретариат ICH, с/о IFPMA, 15 ch. Louis-Dunant, П/Я 195, 1211 Женева 20, Швейцария, http://www.ich.org.
[2] Руководство ICH Q9 Управление рисками по качеству
_______________
Доступно в Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH), Секретариат ICH, с/о IFPMA, 15 ch. Louis-Dunant, П/Я 195, 1211 Женева 20, Швейцария, http://www.ich.org.
[3] Руководство ICH Q10 Фармацевтическая система качества
_______________
Доступно в Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH), Секретариат ICH, с/о IFPMA, 15 ch. Louis-Dunant, П/Я 195, 1211 Женева 20, Швейцария, http://www.ich.org.
[4] Руководство ICH Q11 Разработка и производство лекарственных субстанций (химических и биотехнологических/биологических)
_______________
Доступно в Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека (ICH), Секретариат ICH, с/о IFPMA, 15 ch. Louis-Dunant, П/Я 195, 1211 Женева 20, Швейцария, http://www.ich.org.
[5] АСТМ Е2475 Руководство к пониманию процессов, связанных с фармацевтическим производством и контролем
[6] АСТМ Е2474 Практические подходы к проектированию фармацевтических процессов с использованием процессно-аналитической технологии (ПАТ)
[7] АСТМ Е2537 Руководство по применению непрерывной верификации качества в фармацевтическом и биофармацевтическом производстве
[8] АСТМ Е2629 Руководство по верификации систем контроля с поддержкой процессно-аналитической технологии (ПАТ)
[9] АСТМ Е2476 Руководство по оценке и контролю рисков, влияющих на проектирование, разработку и функционирование ПАТ-процессов в фармацевтическом производстве
Руководство FDA для производителей. Валидация процессов: основные принципы и подходы
_______________
Доступно в Управлении по контролю качества пищевых продуктов и лекарственных средств США (FDA), 5600 Fishers Ln., Роквил, штат Мериленд 20857, http://www.fda.gov.
Современные фармацевтические надлежащие производственные практики (cGMP) XXI века - Подход, основанный на оценке риска
_______________
Доступно в Управлении по контролю качества пищевых продуктов и лекарственных средств США (FDA), 5600 Fishers Ln., Роквил, штат Мериленд 20857, http://www.fda.gov.
УДК 637.132.4:715.478:658.513:006.354 | ОКС 13.060.70 |
Ключевые слова: производство, лекарственные средства, производственные системы, оборудование, общие требования | |
Электронный текст документа
и сверен по:
, 2019
