
Группа НО?
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
ГОСТ
1030-81
Взамен
ГОСТ 1030—41
ВОДА ХОЗЯЙСТВЕННО-ПИТЬЕВОГО НАЗНАЧЕНИЯ
Полевые методы анализа
House keeping and potable water. Field methods of analysis
Постановлением Государственного комитета СССР по стандартам от 11 августа 1981 г. Hs 3765 срок действие установлен
с 01.01.83 до 01.01.88
Несоблюдение стандарта преследуется по закону
Настоящий стандарт распространяется на природные воды хозяйственно-питьевого назначения и устанавливает полевые методы органолептического и физико-химического анализов в процессе гидрогеологической съемки, поисков и разведки источников хозяйственно-питьевого водоснабжения.
Общие требования по ГОСТ 24902—81.
1. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ И ОРГАНОЛЕПТИЧЕСКИХ ПОКАЗАТЕЛЕЙ (ЗАПАХ, ВКУС, ЦВЕТНОСТЬ, МУТНОСТЬ)
1.1. Определение температуры
Температуру воды определяют термометром с делениями на 0, Г С. Для определения температуры воды колодцев и источников применяют родниковые («ленивые1 2) термометры, для воды буровых скважин — максимальные термометры. Помимо ртутных термометров используют также электрические термометры и термоэлементы.
1.2. Определение запаха
В чистый, без запаха стеклянный сосуд вместимостью 50—100 мл наливают анализируемую воду, примерно на 8/< объема, закрывают его стеклянной или корковой пробкой, затем взбалтывают пробу, вынимают пробку и сразу нюхают.
Интенсивности запаха оценивают по пятибалльной системе по ГОСТ 3351—74. При этом учитывают характер запаха (землистый, нефтепродуктов и.др.).
гост teio—st
1.3. Определение вкуса
Вкус воды определяют при отсутствии подозрений на ее загрязненность. Анализируемую воду набирают в рот, не проглатывая, и задерживают несколько секунд. Интенсивность вкуса оценивают по пятибалльной системе по ГОСТ 3351—74. При этом учитывают характер привкуса (соленый, кислый, щелочным, металлический и др.).
1.4. Определение цветности
Цветность определяют только в прозрачной природной воде. Пробирку диаметром 14—16 мм из бесцветного стекла наполняют анализируемой водой до высоты 10—12 см и рассматривают сверху на белом фоне.
Качественно различают следующие степени цветности: бесцветная, слабо-желтоватая, светло-желтая, желтая, интенсивно желтая.
1.5. Определение мутности
Пробирку диаметром 14—16 мм из бесцветного стекла наполняют анализируемой водой до высоты 10—12 см и рассматривают сверху на черном фоне. Качественно различают следующие степени мутности: прозрачная, слабо опалесцирующая, опалесцирующая, слабо мутная, мутная, очень мутная.
2. ОПРЕДЕЛЕНИЕ ХИМИЧЕСКОГО СОСТАВА
2.1. Общие требования
2.1.1. Для анализа химического состава воды применяют: колориметрические методы для определения pH, общего железа, нит-рит-иона, иона аммония, нитрат-иона, фтора и суммы металлов;
объемные методы — для определения карбоната-иона, гидро-карбонат-иона, хлор-иона, иона кальция и общей жесткости;
турбидиметрические методы — для определения сульфат-иона; расчетные методи для определения иона магния, иона натрия, сухого остатка и карбонатной жесткости.
Допускается, кроме установленных настоящим стандартом методов, применять другие методы (например, инструментальные), удовлетворяющие требованиям ГОСТ 24902—81.
2.1.2. Применяемые реактивы должны быть «чистые для анализа» (ч. д. а.) или «химически чистые» (х. ч.), если квалификация специально не оговорена в методе анализа.
2.1.3. Колориметрически^ методы определения компонентов проводят способом «стандартных серий» в пробирках одинакового диаметра из бесцветного стекла с меткой 5 мл. Следует иметь в виду, что возникающие в процессе колориметрических реакций окраски, обычно малоустойчивы. Поэтому шкалы модельных эталонов для сравнения приходится готовить многократно одновременно с проведением анализа воды. Для устранения этого недостатка
разрешается при полевых анализах выполнять колориметрирование не только путем сравнения возникающей окраски с модельными -эталонами, но и с имитирующими эту окраску устойчивыми растворами или окрашенными пленочными шкалами. При работе с пленочными шкалами применяют колориметрические пробирки, внутренний диаметр которых не выходит за пределы (12,8±0,4) мм.
2.1.4. Объемные методы определения компонентов проводят в пробирках вместимостью 15—20 мл и имеющих метку 10 мл. Перед определением пробирку два-три раза ополаскивают анализируемой водой и затем наливают воду до метки. Жидкость перемешивают стеклянной палочкой с шариком на конце. На пробирку с титруемой жидкостью надевают резиновое кольцо и помещают в полевой мутномер (см. п. 2.4.1.4).
Для анализа маломинерализованных вод допускается применять менее концентрированные титрованные растворы (0,02 н.— 0,03 н.).
2.2. Калориметрические методы
2.2.1. Опре деление водородного показателя (pH)
2.2.1.1. Определение выполняют для интервала pH 5,4—8,0 колориметрическим методом, применяя универсальный индикатор.
2.2.1.2. Подготовка к анализу
Приготовление универсального индикатора
Раствор А. 0,100 г индикатора бромтимоловый синий растворяют в фарфоровой ступке в присутствии 8,0 мл 0,02 н. раствора гидроокиси натрия, прибавляют 50 мл этилового спирта ректификата, переносят в мерную колбу вместимостью 250 мл и доводят объем раствора дистиллированной водой до метки.
Раствор Б. 0,025 г индикатора метиловый красный растворяют в фарфоровой ступке в присутствии 4,6 мл 0,02 н. раствора гидроокиси натрия, прибавляют 50 мл этилового спирта ректификата, переносят в мерную колбу вместимостью 250 мл и доводят объем раствора дистиллированной водой до метки.
Растворы А и Б сливают в соотношении 1:1.
Приготовление 0,1%-ного раствора метилового оранжевого — по ГОСТ 4919.1—77.
2.2.1.3. Проведение анализа
Анализируемую воду наливают в колориметрическую пробирку до метки 5 мл, прибавляют при помощи тарированной капельницы 0,10 мл универсального индикатора, перемешивают жидкость и сразу же сравнивают со шкалой эталонов.
Эталоном служат буферные растворы, к 5 мл каждого из которых добавляют 0,10 мл универсального индикатора. Буферные растворы готовят по ГОСТ 4919.2—77.
Шкалу составляют для следующих значений pH: 5,4; 5,6*, 5,8; 6,0; 6,2; 6,4; 6,6; 6,8; 7,0; 7,2; 7,4; 7,6; 7,8; 8,0.
Если pH окажется равным или менее 5,4, анализируемую воду проверяют качественной реакцией, добавляя к 5 мл воды каплю 0,1%-ного раствора метилового оранжевого. Если окраска при этом окажется розовой, записывают результаты: рН<4,5. Анализ пробы на другие компоненты в этом случае прекращают. Если окраска окажется желтой, записывают результат: pH<5,4. Если pH воды окажется больше 8,0, записывают результат: рН>8,0.
2.2.2. Определение массовой концентрации железа (Fe)
Ортофенантролиновый метод
2.2.2.1. Определение основано на способности иона закисного железа образовывать в интервале pH 3—9 с ортофенантролином комплексное соединение, окрашенное в оранжево-красный цвет.
Окисное железо восстанавливают до закисного солянокислым гидроксиламином в нейтральной или слабокислой среде
2.2.2.2. Подготовка к анализу
Приготовление 10%-ного раствора гидроокиси натрия
10 г гидроокиси натрия растворяют в 90 мл дистиллированной воды. Раствор хранят в полиэтиленовом сосуде.
Приготовление 10%-ного раствора солянокислого гидроксиламина.
10 г солянокислого гидроксиламина (NH2OH HC1) растворяют в 90 мл дистиллированной воды.
Приготовление ацетатного буферного раствора
250 г уксуснокислого аммония (CH3COONH4) растворяют в 150 мл дистиллированной воды. Добавляют 700 мл ледяной уксусной кислоты и доводят объем до 1 мл дистиллированной водой. Приготовление 0,1%-ного раствора ортофенантролина
0,10 г моногидрата ортофенантролина (СиоНвМг’НгО) растворяют в 100 мл дистиллированной воды, подкисленной 3—4 каплями соляной кислоты (I : 1). Реактив хранят в темном стеклянном сосуде с притертой пробкой.
2.2.2.3. Проведение анализа
10 мл анализируемой воды в зависимости от pH среды доводят из капельниц 10%-ным раствором гидроокиси натрия или соляной кислотой (1:10) в присутствии индикаторной бумаги конго до перехода окраски бумаги в фиолетовый цвет (рН-4—5). Приливают поочередно 0,2 мл 10%-ного раствора солянокислого гидроксиламина, 1 мл ацетатного буферного раствора и 0,50 мл 0,1%-ного раствора ортофенантролина. После прибавления каждого реактива содержимое пробирки перемешивают. Оставляют раствор не менее чем на 15—20 мин для полного развития окраски. Окрашенный раствор отливают в сухую колориметрическую пробирку до метки 5 мл и сравнивают со шкалой эталонов железа, приготовленных в аналогичных условиях.
Для приготовления шкалы эталонов пользуются разбавленными стандартными растворами соли Мора, приготовленными по ГОСТ 4212—76.
Шкалу составляют для следующих значений Fe (мг/дм3): 0,00; 0,10; 0,20; 0,25; 0,30; 0,40; 0,50; 0,70; 1,00; 1,50.
Если окраска жидкости окажется интенсивнее крайнего эталона (1,5 мг/дм3), анализируемую воду разбавляют б десять раз дистиллированной водой, не содержащей железа, и определение повторяют. При вычислении результатов учитывают величину разбавления.
Дипиридиловыи метод
2.2.2.4. Определение основано на способности иона закисного железа образовывать в интервале pH 3,5—8,5 с а, а'-дипиридилом комплексное соединение, окрашенное в красный цвет.
Окисное железо восстанавливают до закисного солянокислым гидроксиламином в нейтральной или слабокислой среде.
2.2.2.5. Подготовка к анализу
Приготовление 0,1%-ного раствора а, а'-дипиридила
0,10 г реактива растворяют в 100 мл дистиллированной воды.
Реактив хранят в темном стеклянном сосуде с притертой пробкой,.
Приготовление 10%-ного раствора гидроокиси натрия, 10%-ного раствора солянокислого гндроксиламина и ацетатного буферного раствора по п. 2.2.2.2.
2.2.2 6. Проведение анализа
10 мл анализируемой воды в зависимости от pH среды доводят из капельниц 10%-ным раствором гидроокиси натрия или соляной кислотой (I : 10) в присутствии индикаторной бумаги конго до перехода окраски бумаги в фиолетовый цвет (pH = 4—5). Приливают поочередно 0,2 мл 10%-ного раствора солянокислого гидроксиламина, 1 мл ацетатного буферного раствора и 1,0 мл 0,1 %-ного раствора а, а'-дипиридила. После прибавления каждого реактива содержимое пробирки перемешивают. Оставляют6 раствор не менее чем на 15—20 мин для полного развития окраски. Окрашенный раствор отливают в сухую колориметрическую пробирку до метки 5 мл и сравнивают со шкалой эталонов железа, приготовленных в аналогичных условиях.
Для приготовления шкалы эталонов пользуются разбавленными стандартными растворами соли Мора, приготовленными по ГОСТ 4212—76.
Шкалу составляют для следующих значений Fe (мг/дм3): 0,00; 0,10; 0,20; 0,25; 0,30; 0,40; 0,50; 0,70; 1,00; 1,50.
Если окраска жидкости окажется интенсивнее крайнего эталона (1,5 мг/дм3), анализируемую воду разбавляют в десять раз дистиллированной водой, не содержащей железа, и определение повторяют. При вычислении результатов учитывают величину разбавления.
2.2.3. Определение массовой концентрации нитрит- и о н а (NOa-)
2.2.3.1. Определение основано на реакции нитрит-иона с реактивом Грисса. Сульфаниловая кислота и альфа-нафтиламин, содержащиеся в реактиве, образуют в кислой среде с нитрит-ионом окрашенное в розовый цвет азосоединение.
2.2.3.2. Подготовка к анализу
Приготовление реактива Грисса
Тщательно смешивают растертые в ступке до порошкообразного состояния 89 г винно-каменной кислоты, 10 г сульфаниловой кислоты и 1 г альфа-нафтиламина. Герметически закрытый реактив при хранении в темном месте не должен окрашиваться.
2.2.3.3 Проведение анализа
Анализируемую воду наливают в колориметрическую пробирку до метки 5 мл, добавляют около 0,05 г порошка реактива Грисса, раствор взбалтывают до растворения кристаллов и через 15— 20 мин сравнивают со шкалой эталонов азотистокислого натрия, приготовленных в аналогичных условиях. Если температура окружающей среды будет ниже 20—25° С, время выстаивания воды ^увеличивают или подогревают раствор до 50—60° С и через 5— 10 мин колориметрируют.
Для приготовления шкалы эталонов пользуются соответственно разбавленным стандартным раствором азотистокислого натрия, приготовленным по ГОСТ 4212—76.
Шкалу составляют для следующих значений NO2_ (мг/дм3): ОДО; 0,01; 0,02; 0,05; 0,10; 0,20; 0,50; 1,00.
Если окраска жидкости окажется интенсивнее крайнего эталона (1,00 мг/дм3), записывают результат: NO2“ > 1 мг/дм3.
2.2 4. Определение массовой концентрации иона аммония (NH4+)
2.2.4.1. Определение основано на реакции иона аммония с реактивом Несслера. Двуйодистая ртуть, содержащаяся в реактиве, образует в щелочной среде окрашенное в желтый цвет соединение йодистый меркураммоний (0 = Hg2 = NH2 — J).
2.2.4 2, Подготовка к анализу
Приготовление реактива Несслера — по ГОСТ 4517—75. Реактив хранят в полиэтиленовой капельнице.
Приготовление виннокислого калия — натрия (сегнетовая соль) Сегнетовую соль подвергают сушке в термостате при температуре 80—90° С в течение 2—3 ч для удаления аммонийных солей, затем проверяют на чистоту проведением холостого опыта.
2.2.4.3. Проведение анализа
Анализируемую воду наливают в колориметрическую пробирку до метки 5 мл, добавляют ~0,1 г сегнетовой соли и прибавляют из тарированной капельницы 1,0 мл реактива Несслера. Раствор перемешивают и через 1—2 мин сравнивают со шкалой эталонов хлористого аммония, приготовленных в аналогичных условиях.
Для приготовления шкалы, пользуются соответственно разбавленным стандартным раствором хлористого аммония, приготовленным по ГОСТ 4212—76.
Шкалу составляют для следующих значений NH4+ (мг/дм3): 0,0; 0,1; 0,2; 0,4; 0,7; 1,0; 1,5; 2,0; 3,0.
Если окраска жидкости окажется интенсивнее крайнего эталона (3,0 мг/дм3), записывают результат: NH4+>3 мг/дм3.
2.2.5. О п р е д е л е н и е массовой концентрации н-ит-р ат-иона (NO3~)
2.2.5 1. Определение основано на реакции между салициловой кислотой и нитрат-ионами с образованием нитропроизводных салициловой кислоты, которые в щелочной среде окрашены в желтый цвет.
Определению мешает ион хлора, если его массовая концентрация в воде превышает 500 мг/дм3, и железо, если его массовая концентрация превышает 0,5 мг/дм3. От влияния более высоких концентраций железа освобождаются в процессе выполнения анализа. При концентрации хлор-иона более 500 мг/дм3 анализируемую воду разбавляют и определение повторяют.
2.2.5.2 Подготовка к анализу
Приготовление 10%-ного раствора салициловой кислоты или салициловокислого натрия
10 г салициловой кислоты или 10 г салициловокислого натрия растворяют в 90 мл этилового спирта (салициловая кислота) или 90 мл дистиллированной воды (салициловокислый натрий).
Приготовление 20%-ного раствора гидроокиси натрия
20 г гидроокиси натрия растворяют в 80 мл дистиллированной воды. Раствор хранят в полиэтиленовом сосуде плотно закрытым.
2.2.5.3 Проведение анализа
В небольшую фарфоровую чашку наливают пипеткой с делениями на 0,01 см3 1,00 мл анализируемой воды. Если в вбде содержится более 0,5 мг/дм3 железа, в чашку вносят 0,1 г сегнетовой соли. Содержание чашки выпаривают досуха на водяной бане. По охлаждении в чашку добавляют 4—5 капель раствора салициловой кислоты (салициловокислого натрия) так, чтобы смочить весь сухой остаток и осторожно при помощи пипетки с резиновой грушей около 0,5 мл концентрированной серной кислоты плотностью 1,83 г/см3. Стеклянной палочкой тщательно растирают сухой остаток с кислотой по дну и стенкам чашки. Затем, не вынимая палочку из чашки, дают жидкости постоять около 5 мин и добавляют 3—4 мл дистиллированной воды с таким расчетом, чтобы смыть стенки чашки. К полученному сернокислому раствору осторожно приливают маленьким цилиндром или градуированной пробиркой 4—5 мл 20%-ного раствора гидроокиси натрия. При наличии в анализируемой воде нитрат-иона сразу возникает желтая окраска.
Содержимое чашки по стеклянной палочке сливают в пробирку с меткой на 10 мл, ополаскивают чашку и палочку небольшими порциями (по нескольку капель) дистиллированной воды и доводят объем раствора до метки. Если при этом выпадает осадок основных солей магния, то раствор оставляют для отстаивания осадка. Прозрачный раствор наливают в колориметрическую пробирку до метки 5 мл и сравнивают со шкалой эталонов, приготовленных в аналогичных условиях.
Для приготовления шкалы пользуются соответственно разбав-.ленным стандартным раствором азотнокислого калия, приготовленным по ГОСТ 4212—76.
Шкалу составляют для следующих значений NO3- (мг/дм3): 0; 5; 10; 15; 20; 30; 40; 45; 50; 60.
Шкала устойчива в течение 6 мес при хранении в сосудах с притертой стеклянной пробкой в темном месте.
Если окраска жидкости окажется интенсивнее крайнего эталона (60 мг/дм3), анализируемую воду разбавляют в 5 раз дистиллированной водой и определение повторяют. При вычислении результатов учитывают величину разбавления.
2.2.6. Определение массовой концентрации суммы металлов (2Ме)
2.2.6.1. Определение основано на реакции цинка, меди и свинца с дитизоном, в результате которой образуются окрашенные в оранжево-красный цвет дитизонаты металлов.
2.2.6.2. Подготовка к анализу
Приготовление аммиака
Для определения суммы металлов применяют аммиак высокой степени чистоты. При отсутствии аммиака квалификации «ос. ч», его получают насыщением очищенной дистиллированной воды концентрированным аммиаком. Для этого в эксикатор наливают 1 л 25%-ного аммиака и на вкладыш эксикатора ставят выпарительную чашку с 500 мл очищенной воды. Эксикатор закрывают и оставляют на двое суток. Получают в чашке аммиак концентрацией ~ 17%.
Очищенную дистиллированную воду получают повторной перегонкой дистиллированной воды. Воду доводят до необходимой чистоты 0,01%-ным раствором очищенного дитизона в четыреххлористом углероде, добавляя на 300—500 мл воды порциями по Ю—15 мл дитизона до тех пор, пока зеленый цвет раствора перестанет изменяться.
Полученный очищенный раствор аммиака разбавляют очищенной дистиллированной водой в 10 раз, прибавляя 10 мл аммиака к 100 мл воды.
Приготовление буферного раствора (рН = 8,0)
Смешивают 55,9 мл 0,05 н. раствора буры с 44,1 мл 0,1 н. раствора соляной кислоты (см. ГОСТ 4819.2—77). Компоненты буферных растворов готовят на очищенной дистиллированной воде.
Приготовление четыреххлористого углерода
Четыреххлористый углерод очищают перегонкой в стеклянном аппарате с дефлегматором при 76° С на водяной бане. Перегонку проводят под тягой.
Приготовление очищенного дитизона
1 г препарата растворяют в 100 мл хлороформа. Жидкость помещают в делительную воронку вместимостью 500 мл, добавляют 10 мл 3%-ного раствора аскорбиновой кислоты и 100 мл разбавленного очищенного аммиака (1 : 100). Встряхивают смесь в воронке в течение 2 мин, затем оставляют воронку в вертикальном положении до полного разделения слоев. Нижний хлороформенный слой сливают в другую делительную воронку, следя за тем, чтобы в оранжевом водном аммиачном растворе не осталось капелек хлороформа. Извлечение дитизона свежими порциями аммиачного раствора с аскорбиновой кислотой повторяют до тех пор, пока новые порции водно-аммиачного раствора перестанут окрашиваться ь желтый цвет (для этого обычно требуется 5—6 извлечений). Аммиачные экстракты^ содержащие дитизон, собирают вместе в делительную воронку вместимостью 1 л и, осторожно помешивая, нейтрализуют соляной кислотой (1 : 1), пока дитизон выпадет в виде темных хлопьев, а цвет раствора из оранжевого перейдет в бледно-зеленоватый. Полученный дитизон отфильтровывают через бумажный фильтр, два-три раза промывают 1%-ным водным раствором аскорбиновой кислоты, собирая осадок струей из про-мывалки в нижнюю часть фильтра, и оставляют на воздухе до высушивания. Очищенный дитизон хранят в темной бюксе или пробирке с притертой пробкой. Все работы по очистке дитизона проводят в вытяжном шкафу.
Приготовление 0,01%-ного раствора дитизона
0,010 г дитизона растворяют в 100 мл четыреххлористого углерода. Раствор хранят в темном стеклянном сосуде с притертой пробкой. Срок годности 6 мес.
Чистота применяемых для определения суммы -металлов реактивов и посуды проверяется проведением холостых опытов.
2.2.6.3. Проведение анализа
В ополоснутую несколько раз анализируемой водой делительную воронку вместимостью 100 мл с предварительно нанесенной на ней меткой 25 мл наливают до метки анализируемую воду, прибавляют 1,0 мл буферного раствора п при помощи тарированной
ГОСТ 1030—ei
капельницы или пипетки с резиновой грушей 2,0 мл 0,01 %-ного раствора дитизона. Встряхивают содержимое воронки в течение 1 мин, вносят из капельницы 2 капли раствора очищенного аммиака (концентрации 1:10) и вновь встряхивают в течение 15— 20 с. Воронку оставляют до расслоения жидкости. После расслоения сливают органический слой в колориметрическую пробирку и сравнивают со шкалой эталонов, приготовленных в тех же условиях.
Основной стандартный раствор суммы металлов готовят из смеси цинка, меди и свинца в молярных соотношениях 3:1:1. С этой целью в мерную колбу вместимостью 1 л помещают 0,392 г металлического цинка, 0,127 г металлической меди и 0,414 г металлического свинца, приливают 20 мл концентрированной азотной кислоты и после растворения металлов доливают дистиллированной водой до метки. Получают 0,01 М раствор суммы металлов.
Раствор устойчив в течение года.
Из основного стандартного раствора готовят рабочий стандартный раствор концентрацией 0,001 ммоль/дм3.
Разбавление проводят в два этапа. Вначале получают промежуточный раствор разбавлением основного раствора в 100 раз, а затем разбавлением промежуточного раствора еще в 100 раз.
Промежуточный раствор устойчив в течение месяца.
Из рабочего стандартного раствора соответствующим разбавлением приготавливают эталоны шкалы. Все операции по приготовлению конечного рабочего и эталонных растворов выполняются в день проведения анализа.
Все разбавления проводят очищенной дистиллированной водой. Шкалу составляют для следующих значений в анализируемой воде SMe: 0,0000; 0,0001; 0,0002; 0,0003; 0,0005; 0,0008;
0,0010 ммоль/дм3, что соответствует в условном пересчете 2Ме на цинк (с округлением до одного значащего десятичного знака) 0,000; 0,006; 0,010; 0,020; 0,030; 0,050; 0,060 мг/дм3.
Если окраска жидкости окажется интенсивнее крайнего эталона, результат записывают: SMe>0,001 ммоль/дм3.
2.2.7. О п р е д е л е н и е массовой концентрации фтора (F)
2.2.7.1. Определение основано на реакции фтора и лантанали-заринкомплексонового лака с образованием окрашенного в синий цвет тройного комплекса фторида, трехвалентного лантана и али-заринкомплексона. Определению мешают алюминий, железо и повышенное содержание органических веществ.
Ввиду того, что содержание алюминия в нейтральных и около-нейтральных природных водах (рН = 6—8) обычно очень незначительно, влиянием алюминия пренебрегают.
От влияния органических веществ освобождаются, как указано в п. 2.2.7.3.
Железо начинает оказывать заметное влияние на определение фтора при массовой концентрации железа более 2 мг/дм3. Поэтому в сильно железистых водах определение фтора по настоящему стандарту не проводят.
2.2.7.2. Подготовка к анализу
Приготовление лантанализаринкомплексонового лака
Лак готовят смешиванием следующих растворов
Раствор соли лантана: 1,293 г бромистого лантана (ЬаВгзХ Х7Н2О) или 0,905 г хлористого лантана (LaCI3-6H2O) помещают в мерную колбу вместимостью 100 мл, приливают 10 мл кислоты (1:10) и доводят дистиллированной водой до метки;
раствор ализаринкомплексона: 0,240 г ализаринкомплексона помещают в небольшой стаканчик или стеклянную бюксу, приливают 11 мл аммиачного буферного раствора и растирают стеклянной палочкой до полного растворения реактива;
аммиачный буферный раствор: 20 г уксуснокислого аммония (CH3COONH4) растворяют в мерной колбе вместимостью 100 мл в 30—50 мл дистиллированной воды, приливают 10 мл 25%-ного раствора аммиака и доводят до метки дистиллированной водой;
ацетатный буферный раствор (рН = 5,0):41 г уксуснокислого аммония (CH3COONH4) помещают в мерную колбу вместимостью 250 мл; смачивают дистиллированной водой, приливают 30 мл ледяной уксусной кислоты и доводят до метки дистиллированной водой.
Лантанализаринкомплексоновый лак готовят из указанных растворов следующим образом: в мерную колбу вместимостью 1 л наливают 50,0 мл раствора соли лантана, прибавляют весь объем (250 мл) ацетатного буферного раствора и весь объем (11,0 мл) раствора ализаринкомплексона, приливают 500 мл ацетона и доводят дистиллированной водой до метки. Ввиду токсичности и горючести ацетона реактив готовят под тягой.
Раствор хранят в склянке из стекла с притертой пробкой в темном месте. Срок годности 6 мес.
Приготовление буферной смеси
3,60 г янтарной кислоты растирают в ступке с 7,00 г тетра-борнокислого натрия (Na2B4O7* ЮН2О). Смесь хранят в плотно закрытом сосуде.
2.2.7.3. Проведение анализа
Анализируемую воду наливают в колориметрическую пробирку до метки 5 мл, вносят ~0,1 г буферной смеси и перемешивают до растворения; pH раствора при этом будет ~ 5,0.
Затем приливают к пробе при помощи пипетки с резиновой грушей 2,0 мл лантанализаринкомплексонового лака, перемешивают и через 20 мин сравнивают со шкалой эталонов, приготовленных в аналогичных условиях.
ГОСТ 1030—31
Для приготовления шкалы эталонов пользуются разбавленным стандартным раствором фтористого натрия, приготовленным по ГОСТ 4212—76.
Шкалу составляют для следующих значений F (мг/дм3): 0,0; 0,5; 0,7; 1,0; 1,2; 1,5; 1,7; 2,0.
Если окраска жидкости окажется синее, чем крайний эталон (2,0 мг/дм3), анализируемую воду разбавляют в 2—5 раз дистиллированной водой и определение повторяют. При вычислении результатов учитывают величину разбавления.
При повышенном содержании в анализируемой воде растворенных органических веществ определить фтор по настоящему стандарту невозможно (окраска колориметрируемой жидкости получается другого оттенка, чем шкала эталонов). В этих случаях порцию анализируемой воды объемом 20—25 мл встряхивают в течение 3—5 мин с небольшим количеством порошка активированного угля марки БАУ или ОУ щелочной и фильтруют. Определение фтора выполняют из фильтрата.
2.3. Объемные методы
2.3.1. Определение массовой концентрации кар-бонат-иона (СОз2^)
2.3.1.1. Определение основано на реакции карбонат-иона с ионами водорода:
СО|~ + Н+-> нсо-.
Анализ проводится титриметрическим методом в присутствии индикатора фенолфталеина. Присутствие аналитических концентраций карбонат-иона возможно лишь в водах, pH которых более 8,0—8,2.
2.3.1.2. Подготовка к анализу
Приготовление 0,1%-ного раствора фенолфталеина — по ГОСТ 4919.1—77.
Приготовление 0,05 н. титрованного раствора соляной кислоты из фиксанала.
2.3.1.3. Проведение анализа
В пробирку с меткой 10 мл наливают до метки анализируемую воду и прибавляют из тарированной капельницы ~0,1 мл 0,1 %-ного раствора фенолфталеина. Если вода остается бесцветной или слабо-розовой — карбонат-ион отсутствует. Если жидкость окрасится в отчетливо розовый цвет, ее титруют 0,05 н. раствором соляной кислоты до тех пор, пока окраска побледнеет до слаборозовой. Полученный раствор оставляют для определения гидро-карбонат-иона (см. п. 2.3.2.3).
2.3.1.4. Обработка результатов
Массовую концентрацию карбонат-иона (X) в мг/дм3 вычисляют по формуле
V • н • 60 . 1000
х = Vl
где V — объем титрованного раствора соляной кислоты, израсходованный на определение, мл;
н — нормальность раствора соляной кислоты;
Vi — объем воды, взятый для анализа, мл;
60 — эквивалентная масса карбонат-иона в данной реакции.
Полученный результат округляют до целых чисел.
2.3.2. О п р ед ел е н и е массовой концентрации гид-рокарбонат-иона (НСОз“)
2.3.2.1. Определение основано на реакции гидрокарбонат-иона с ионами водорода:
НСО^ + Н ’ ->СО2+Н2О.
Анализ проводится титриметрическим методом в присутствии индикатора метилового оранжевого.
2.3.2.2. Подготовка к анализу
Приготовление 0,1 % -ного раствора метилового оранжевого — по ГОСТ 4919.1—77.
Приготовление 0,05 н. титрованного раствора соляной кислоты из фиксанала
2.3.2.3. Проведение анализа
В пробирку с меткой 10 мл наливают до метки анализируемую воду и прибавляют 1—2 капли 0,1%-ного раствора метилового оранжевого. Титруют жидкость 0,05 и. раствором соляной кислоты до перехода желтой окраски жидкости в розовую.
Если в анализируемой воде был найден карбонат-ион, определение гидрокарбонат-иона продолжают в той же пробе, в которой определялся карбонат-ион (см. п. 2.3.1.3). При вычислении массовой концентрации количество титрованного раствора соляной кислоты уменьшают на объем, пошедший при определении карбонат-иона.
2.3.2.4. Обработка результатов
Массовую концентрацию гидрокарбонат-иона (А\) в мг/дм3 вычисляют по формуле
V - У3) • н . 61 . 1000
д--------------
где Vz — объем титрованного раствора соляной кислоты, израсходованный на определение НСОз", мл;
У3 — объем титрованного раствора соляной кислоты, израсходованный на определение СО32_, мл;
н — нормальность раствора соляной кислоты;
У4— объем воды, взятый на анализ, мл;
61 —эквивалентная масса гидрокарбонат иона.
Полученный результат округляют до целых чисел.
2.3.3. Определение массовой концентрации хлор-иона (С1“)
Меркуриметрический метод
2.3.3.1. Определение основано на реакции хлор-иона с ионом двухвалентной ртути, которая образует растворимую, но мало-диссоциированную соль — хлорную ртуть:
Hg2- + 2CI'^HgCl2.
В качестве индикатора используется дифенилкарбазон. Величина pH титруемого раствора устанавливается в интервале 3,0—3,5 (контролируется индикатором бромфеноловым синим в процессе проведения анализа).
2.3.3.2. Подготовка к анализу
Приготовление смешанного индикатора (раствор)
0,50 г дифенилкарбазона и 0,050 г бромфенолового синего растворяют в 100 мл спирта ректификата.
Приготовление азотной кислоты (1:200)
Приливают мерным цилиндром 5 мл концентрированной азотной кислоты к 1 л дистиллированной воды.
Приготовление азотнокислой ртути 0,05 н. титрованного раствора.
8,4 г азотнокислой ртути (Hg(NO3)2- 1/2Н2О) растворяют в 100 мл дистиллированной воды, содержащей 1—1,5 мл концентрированной азотной кислоты, и доводят раствор дистиллированной водой до 1 л. Нормальность раствора азотнокислой ртути устанавливают по 0,05 н. раствору хлористого натрия, проводят титрование по п. 2.3.3.3.
2.3.3.3. Проведение анализа
В пробирку с меткой 10 мл наливают до метки анализируемую воду и прибавляют из тарированной капельницы около 0,1 мл раствора смешанного индикатора до фиолетовой окраски. Прибавляют по каплям раствор азотной кислоты (1:200) до перехода окраски в желтый цвет и еще 1—2 капли азотной кислоты. Подготовленную таким образом жидкость титруют 0,05 н. раствором азотнокислой ртути до перехода окраски из желтой в сине-фиолетовую.
2.3.3.4. Обработка результатов
Массовую концентрацию хлор-иона (Хг) в мг/дм3 вычисляют по формуле
v Р5. н- 35.5. 1000
2
где Vs— объем титрованного раствора азотнокислой ртути, израсходованный на определение, мл;
н — нормальность титрованного раствора азотнокислой ртути;
Уб — объем воды, взятый на анализ, мл;
35,5 — эквивалентная масса хлора.
Полученный результат округляют до целых чисел.
Аргентометрический метод
2.3.3.5. Метод основан на реакции хлор-иона с ионом серебра:
Ag* +Cl-~>Ag Cl 1.
В качестве индикатора используется хромовокислый калий.
Титрование можно выполнять в пределах pH (5,0—8,0).
2.3.3.6. Подготовка к анализу
Приготовление 10%-ного раствора хромовокислого калия
10 г К2СГО4 растворяют в 90 мл дистиллированной воды.
Приготовление 0,05 н. титрованного раствора азотнокислого серебра
Готовят из фиксанала или из препарата соли азотнокислого серебра. В последнем случае навеску 8,5 г AgNO3 растворяют в 1 л дистиллированной воды и устанавливают нормальность по 0,05—0,1-н. раствору хлористого натрия, приготовленному из фиксанала. Титрование проводят по п. 2.3.3.3.
2.3.3.7. Проведение анализа
В пробирку с меткой 10 мл наливают до метки анализируемую воду, прибавляют из тарированной капельницы -—0,1 мл 10%-но-го раствора хромовокислого калия и титруют 0,05 н. раствором азотнокислого серебра до появления неисчезающей бурой окраски.
2.3.3.8. Обработка результатов
Массовую концентрацию хлор-иона (Хг) в мг/дм3 вычисляют по п. 2.3.3.4.
2.3.4. Определение общей жесткости
2.3.4.1. Определение основано на реакции солей кальция и магния с реактивом двунатриевая соль этилендиаминтетрауксусной кислоты (трилон Б):
Са2+ + Na2HaR Na2CaR 4- 2Н+;
Mg2+ 4- Na2H2R Na2MgR 4- 2Н+,
где /?— радикал этилендиаминтетрауксусной кислоты.
Анализ проводится титриметрическим методом в присутствии индикатора эриохромчерный Т.
2.3.4.2. Подготовка к анализу
Приготовление буферного раствора
5 г хлористого аммония (NH4C1) растворяют в дистиллированной воде, добавляют 40 мл 10%-ного раствора аммиака, приготовленного по ГОСТ 4517—75, и доводят объем дистиллированной водой до 100 мл.
Приготовление индикатора эриохромчерный Т по ГОСТ 4919.1—77.
Приготовление 0,05 н. титрованного раствора трилона Б
из фиксанала
2.3.4.3. Проведение анализа
В пробирку с меткой 10 мл наливают до метки анализируемую воду из тарированной капельницы, приливают 0,2 мл буферного раствора и вносят 0,02—0,03 г смеси индикатора эриохромчерный Т. Раствор перемешивают и медленно титруют 0,05 н. раствором трилона Б до перехода окраски из винно-красной через фиолетово-синюю в ярко-голубую.
2.3.4.4. Обработка результатов
Общую жесткость (Х3) в мг-экв/дм3 вычисляют по формуле
у У7 - и- 1000
3“ И8
где V7 — объем титрованного раствора трилона Б, израсходованный на определение, мл;
н — нормальность раствора трилона Б;
— объем воды, взятый на анализ, мл.
Полученный результат округляют до второго десятичного знака.
2.3.5. Определение массовой концентрации иона кальция (Са2+)
2.3.5.1. Определение основано на реакции иона кальция с реак
тивом двунатриевая соль этилендиаминтетрауксусной кислоты (трилон Б): Са2+ 4- Na2H2R -> Na2CaR + 2Н+,
где R — радикал этилендиаминтетрауксусной кислоты.
Анализ проводится титриметрическим методом в сильно щелочной среде (pH ~ 12—13) в присутствии индикатора флуорексона (кальцеин).
2.3.5.2. Подготовка к анализу
Приготовление флуорексона (кальцеина) —по ГОСТ 4919.1—77.
Приготовление соляной кислоты (1:100)
Приливают мерным цилиндриком 10 мл концентрированной соляной кислоты (плотность 1,19 г/см3) к 1 мл дистиллированной воды.
Приготовление 10%-ного раствора гидроокиси натрия
10 г гидроокиси натрия растворяют в 90 мл дистиллированной воды. Раствор отстаивают 2 дня и хранят в плотно закрытом полиэтиленовом сосуде.
Приготовление 0,05 н. титрованного раствора трилона Б из фиксанала
2.3.5.3. Проведение анализа
В пробирку с меткой 10 мл наливают до метки анализируемую воду и удаляют гидрокарбонат-ион. Для этого в пробирку помещают кусочек бумаги конго и прибавляют по каплям раствор соляной кислоты (1 : 100) при интенсивном перемешивании шариком-мешалкой до перехода окраски бумаги из красной в сиреневую. При перемешивании удаляется и большая часть двуокиси углерода, мешающего определению.
Бумагу извлекают и прибавляют из тарированной полиэтиленовой капельницы ~0,5 мл 10%-ного раствора гидроокиси натрия и 0,02—0,03 г индикатора флуорексон (индикатор целесообразно отбирать титрованным мерничком). Раствор перемешивают и титруют на фоне черной бумаги 0,05 н. раствором трилона Б до перехода флуоресцирующей окраски из зеленой в розовую.
2.3.5.4. Обработка результатов
Массовую концентрацию иона кальция (Х4) в мг/дм3 вычисляют по формуле
у _ У, ■ н * 20• 1000
где Уд — объем титрованного раствора трилона Б, израсходованный на определение, мл;
н — нормальность раствора трилона Б;
Ую— объемы воды, взятой на анализ, мл;
20 — эквивалентная масса кальция.
Полученный результат округляют до целых чисел.
2.4. Турбидиметрические методы
2.4.1. Определение массовой концентрации сульфонат-иона (SO42”)
2.4.1.1. Определение основано на реакции сульфат-иона с ионами бария и образовании осадка сульфата бария.
SO2" + Ва2+ BaSO4 4 .
Для определения величины Осадка используются различные варианты турбидиметрического метода.
В водах, содержащих SO4^ менее 30 мг/дма, определение проводят путем сравнения образовавшегося осадка с эталонами или со «стандартной суспензией», а при содержании более 30 мг/дм3 — по измерению высоты столба суспензии сульфата бария.
Анализ выполняют в прозрачной воде. Если вода содержит механические примеси и взвеси, ес предварительно фильтруют.
Метод определения сравнения с эталонами
2.4.1.2. Определение основано на сравнении интенсивности образовавшегося помутнения -от выделившегося осадка сульфата бария с модельными эталонами, приготовленными одновременно с анализируемой пробой.
2.4.1.3. Подготовка к анализу
Приготовление азотнокислого бария (насыщенный раствор)
10 г Ba (NO3)2 растворяют при нагревании в 100 мл дистиллированной воды и охлаждают до комнатной температуры. Выпавшие кристаллы азотнокислого бария не препятствуют использованию раствора.
2.4.1.4. Проведение анализа
Анализируемую воду наливают в пробирку до метки 10 мл, добавляют из капельницы 2 капли раствора соляной кислоты (1:1) и 0,5 мл насыщенного раствора азотнокислого бария, взбалтывают и через 10—15 мин сравнивают со шкалой эталонов, приготовленных в аналогичных условиях в пробирках того же диаметра и цвета стекла.
Шкалу рекомендуется составить для следующих значений SO4^ (мг/дм3): 0; 5; 10; 20; 25; 30.
Метод определения с применением стандартной суспензии
2.4.1.5. Определение основано на сопоставлении интенсивности образовавшегося помутнения от выделившегося осадка сульфата бария, с помутнением, образующимся при внесении в воду, помещенную в контрольную пробирку, суспензии сульфата бария.
2.4.1.6. Подготовка к анализу
Приготовление азотнокислого бария (насыщенный раствор) и соляной кислоты (1:1) по п. 2.4.1.3.
Приготовление стандартной суспензии
Стандартную суспензию готовят смешиванием двух растворов.
Раствор А—1,09 г высушенного при температуре 105—110° С Ba (NO3)2, растворяют в мерной колбе вместимостью 1 л в дистиллированной воде.
Раствор Б — 0,1 н. (приблизительно) раствор серной кислоты, в которую добавляют 50 мл 5%-ного раствора питьевого желатина на каждый литр кислоты (5 г желатина растворяют в 95 мл горячей воды).
Стандартную суспензию готовят смешиванием равных объемов растворов А и Б и через 10—15 мин употребляют. 1 мл такой суспензии содержит 0,486 мг BaSO4, что соответствует 0,200 мг SO^. Суспензия пригодна в течение рабочего дня.
2.4.1.7. Проведение анализа
Анализируемую воду наливают в пробирку из бесцветного стекла до метки 10 мл, добавляют из капельницы 2 капли раствора соляной кислоты (1:1) и 0,5 мл насыщенного раствора азотнокислого бария и взбалтывают. Через 10—15 мин перемешивают еще раз. Одновременно с последним перемешиванием в другую пробирку такого же диаметра наливают 10 мл той же анализируемой воды и при помощи градуировочной пипетки приливают хорошо взболтанную свежеприготовленную «стандартную суспензиюэ до образования помутнения, одинакового по интенсивности с помутнением в первой пробирке.
2.4.1.8. Обработка результатов
Массовую концентрацию сульфат-иона (Х5) в мг/дм3 вычисляют по формуле
Х5 - . 0,2 . 100,
где Vh — объем стандартной суспензии, мл;
0,2 — концентрация суспензии, соответствующая мг/см3;
100 — коэффициент пересчета на объем 1 л.
2.4.1.9. Метод определения по высоте столба суспензии
Определение основано на изменении степени помутнения от осадка сульфата бария, образующегося в результате реакции сульфат-иона и иона бария при соответствующих условиях проведения анализа.
2.4.1.10. Аппаратура .
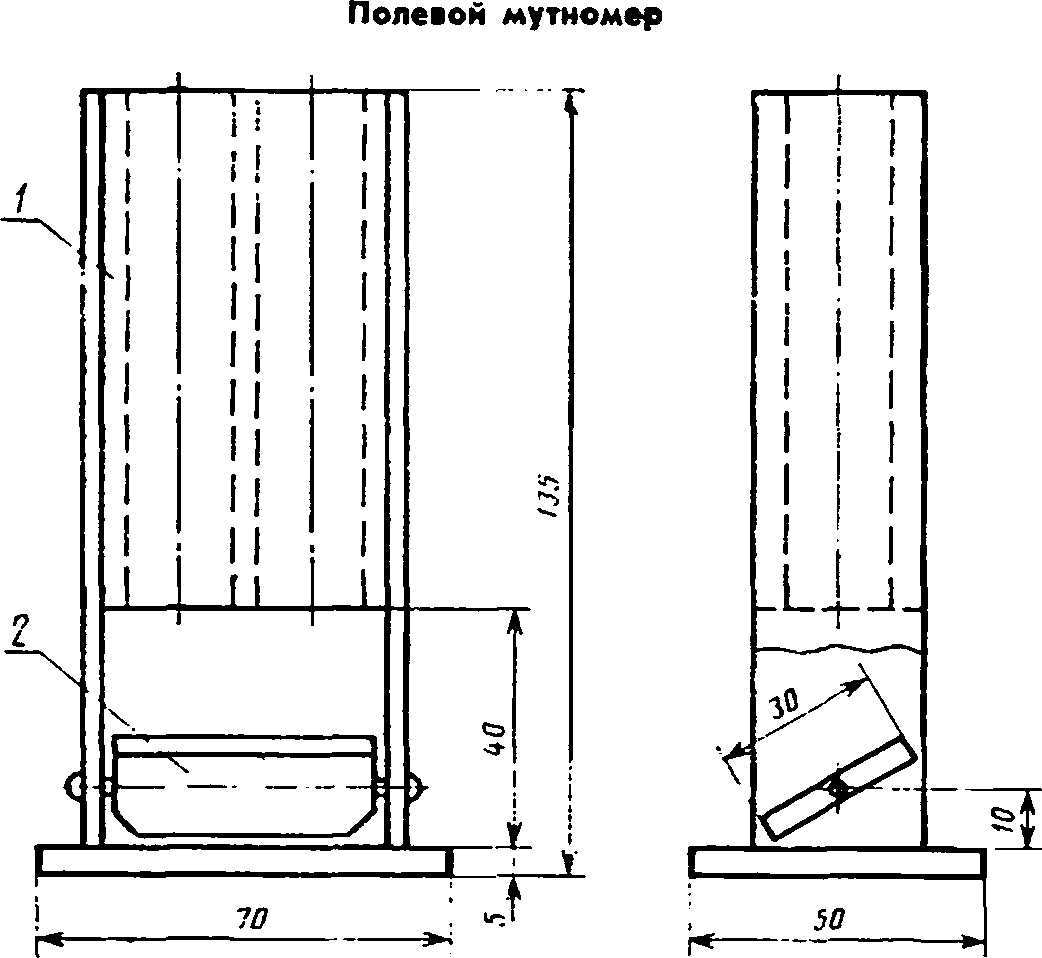
Пробирки мутномерные. Применяются пробирки бесцветного прозрачного стекла внутренним диаметром 14,0—14,5 мм и вместимостью около 20 мл. Пробирки градуируются по высоте до 100 мм через I мм (градуировка может быть заменена полоской миллиметровой бумаги, наклеенной на пробирку). На дне пробирки черным лаком или другой несмывающейся краской наносят по всей площади дна крест с четырьмя точками. Толщина линий креста и точек около 1 мм.
Мутномер полевой. Полевой мутномер (см. чертеж) представляет собой брусок, выполненный из дерева или темной пластмассы с двумя просверленными сквозными отверстиями для мутномерных пробирок. Он укрепляется на плоской фанерной или пластмассовой подставке. Нижняя часть бруска обрезана на расстоянии 40 мм от подставки. В обрезанной части бруска к стенкам мутномера крепится экран, представляющий собой дощечку с белой поверхностью. Экран должен иметь возможность вращаться на 45°.
2.4.1.11. Подготовка к анализу
Приготовление азотнокислого бария (насыщенный раствор) и соляной кислоты (1:1) по п. 2.4.1.3.
2.4. L. 12. Проведение анализа
Для проведения анализа пользуются двумя мутномерными пробирками. На каждую пробирку надевают резиновое кольцо и обе пробирки вставляют в отверстия мутномера. Передвижением колец пробирки фиксируют таким образом, чтобы нижняя часть пробирок была выдвинута в вырез мутномера на расстояние около 1 см. Экран устанавливают под углом 45° к подставке* (при этом дно пробирок окажется на требуемом расстоянии 2 см от экрана).
Работа проводится при рассеянном, но достаточно сильном (200—500 лк) дневном освещении экрана.
В одну из мутномерных пробирок наливают анализируемую воду до высоты 100 мм. Прибавляют из капельницы 2 капли раст-
ГОСТ 1030—01
2 сгт>5 QfS
\ | ||||||
» 1 | ||||||
кг? - 5J | ► | |||||
/ — отверстие для мутномерных пробирок; 2 — экран
вора соляной кислоты (1:1) и 0,5 мл насыщенного раствора азотнокислого бария. Содержимое пробирки взбалтывают и оставляют на 5—7 мин, после чего пробирку взбалтывают еще раз и образовавшуюся суспензию BaSO< отбирают пипеткой в другую пустую мутномерную пробирку до тех пор, пока в первой пробирке появится едва заметное изображение точек рисунка на дне и затем измеряют высоту столба оставшейся суспензии, которую контролируют в другой пробирке, отмечая момент, когда изображение 230
гост изо—ai
точек на ее дне скроется. Берут средний результат обоих измерений и по табл. 3 находят содержание сульфат-иона в анализируемой воде.
Таблица 3
Высота столба суспензии, мм | Массовая концентрация SO4 , мг/дм’ | N Высота столба J суспензии, мм | Массовая концентра- 2— цня SO 4 , мг/дм* |
100 | 33 | 65 | 50 |
95 | 35 | 60 | 53 |
90 | 38 | 55 | 56 |
85 | 40 | 50 | 59 |
80 | 42 | 45 | 64 |
75 | 45 | 40 | 72 |
70 | 47 | I 1 |
Если изображение на дне мутномерной пробирки при высоте столба суспензии окажется менее 40 мм, пробу разбавляют дистиллированной водой в два раза и определение повторяют.
Если в этом случае суспензия окажется слишком плотной, повторяют определение при разбавлении анализируемой воды в 4 раза и т. д., увеличивая разбавление каждый раз вдвое. Общую величину разбавления учитывают при вычислении окончательного результата.
2.5. Расчетные методы
2.5.1. Определение массовой концентрации иона магния (Mg2*)
Массовую концентрацию иона магния (Хе) в мг/дм3 вычисляют по формуле
Х6 = (а — с-0,05). 12,16,
где а — общая жесткость, мг/дм3;
с — массовая концентрация нона кальция, мг/дм3;
0,05 — коэффициент для пересчета нона кальция в миллиграмм-эквивалентную форму;
12,16 — эквивалентная масса магния.
Полученный результат округляют до целых чисел.
2.5.2. Определение массовой концентрации иона натрия (Na+)
Массовую концентрацию иона натрия (Х7) в мг/дм3 вычисляют по формуле
Х7 = (S4 — а) • 23,
где S4—сумма анионов, мг-экв/дм3 (см. табл. 4);
а — общая жесткость, мг-экв/дм3;
23 — эквивалентная масса натрия.
ГОСТ 1030—81
Ввиду того» что массовая концентрация иона калия в природных водах обычно играет подчиненную роль, ее условно учитывают в данном расчете в виде массовой концентрации иона натрия.
Полученный результат округляют до целых чисел.
Для пересчета массовых концентраций ионов в миллиграмм-эквивалентную форму массовые концентрации главных ионов, найденные анализом и выраженные в мг/дм3, умножают на коэффициенты, указанные в табл. 4.
Таблица 4
Анноны | Коэффициент | Катионы | Коэффициент |
НСО^" | 0,0164 | Са2+ | 0,0500 |
СО 23- | 0,0333 | Mg2+ | 0,0822 |
Cl- | 0,0282 | ||
sol" | 0,0208 | ||
NO Г | 0.0161 |
2.5.3. Определение сухого остатка
Результаты определения массовых концентраций всех ионов, выраженные в мг/дм3, суммируют (гидрокарбонат-ион суммируется в количестве 50%). Получают вычисленный сухой остаток, выраженный в мг/дм3.
2.5.4. Вычисление карбонатной жесткости
Карбонатную жесткость вычисляют в мг-экв/дм3 путем суммирования гидрокарбонат и карбонат-ионов, выраженных в мг-экв/дм3.
Если карбонатная жесткость окажется больше общей жесткости, ее считают равной последней.
232
1
Издание официальное Перепечатка воспрещена
2
