
ГОСТ Р 55038-2012
(ИСО 16672:2003)
Группа Р22
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Оптика офтальмологическая
ИМПЛАНТАТЫ ОФТАЛЬМОЛОГИЧЕСКИЕ. ЭНДОТАМПОНАДЫ ГЛАЗНЫЕ
Общие требования безопасности
Ophthalmic optics. Ophthalmic implants. Ocular endotamponades. General safety requirements
ОКС 11.040.40
ОКП 94 3800
Дата введения 2014-07-01
Предисловие
1 ПОДГОТОВЛЕН Открытым акционерным обществом "ТКС-оптика" совместно с рабочей группой ПК 7 "Офтальмологическая оптика" Технического комитета ТК 296 "Оптика и оптические приборы" на основе собственного аутентичного перевода на русский язык международного стандарта, указанного в пункте 4
2 ВНЕСЕН Управлением технического регулирования и стандартизации Федерального агентства по техническому регулированию и метрологии
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 01 ноября 2012 г. N 685-ст
4 Настоящий стандарт является модифицированным по отношению к международному стандарту ИСО 16672:2003* "Имплантаты офтальмологические. Эндотампонады глазные" (ISO 16672:2003 "Ophthalmic implants - Ocular endotamponades") путем:
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
- изменения наименования. Наименование настоящего стандарта изменено относительно наименования указанного международного стандарта для приведения его в соответствие с ГОСТ Р 1.5-2004 (подраздел 3.5);
- изменения структуры. Сравнение структуры настоящего стандарта со структурой указанного международного стандарта приведено в дополнительном приложении ДА;
- введения дополнительных терминов с соответствующими определениями для лучшего понимания текста стандарта, которые в тексте стандарта выделены курсивом*;
________________
* В бумажном оригинале обозначения и номера стандартов и нормативных документов в разделе "Предисловие", приложениях ДА, ДБ приводятся обычным шрифтом, отмеченные в этих разделах знаком "**" и остальные по тексту документа выделены курсивом. - .
- изменения отдельных фраз, которые в тексте стандарта также выделены курсивом;
- введения дополнительных положений, которые в тексте стандарта также выделены курсивом. Внесение указанных дополнительных положений обусловлено особенностью объекта стандартизации, характерной для Российской Федерации.
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации и межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДБ
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012** (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (gost.ru)
1 Область применения
Настоящий стандарт распространяется на глазные эндотампонады (далее - ГЭ) класса неактивных хирургических имплантатов, используемые в офтальмологии. ГЭ предназначены для сглаживания и закрепления положения отслоившейся сетчатки на сосудистой оболочке или тампонирования сетчатки.
Стандарт устанавливает требования безопасности в отношении предусмотренного применения, характеристик конструкции, доклинической и клинической оценок, стерилизации, упаковки и маркировки изделия и информации, предоставляемой изготовителем этих изделий.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты*:
________________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ГОСТ Р ИСО 10993-1-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 1. Оценка и исследования
ГОСТ Р ИСО 10993-2-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 2. Требования к обращению с животными
ГОСТ Р ИСО 10993-3-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 3. Исследование генотоксичности, канцерогенности и токсического действия на репродуктивную функцию
ГОСТ Р ИСО 10993-5-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 5. Исследования на цитотоксичность: методы in vitro
ГОСТ Р ИСО 10993-6-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 6. Исследование местного действия после имплантации
ГОСТ Р ИСО 10993-7-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 7. Остаточное содержание этиленоксида после стерилизации
ГОСТ Р ИСО 10993-10-2009 Изделия медицинские. Оценка биологического действия медицинских изделий. Часть 10. Исследование раздражающего и сенсибилизирующего действия
ГОСТ Р ИСО 11134-2000 Стерилизация медицинской продукции. Требования к валидации и текущему контролю. Промышленная стерилизация влажным теплом
ГОСТ Р ИСО 11135-2000 Медицинские изделия. Валидация и текущий контроль стерилизации оксидом этилена
ГОСТ Р ИСО 13408-1-2011* Асептическое производство медицинской продукции. Часть 1. Общие требования
________________
* Вероятно, ошибка оригинала. Следует читать: ГОСТ Р ИСО 13408-1-2000, здесь и далее по тексту. - .
ГОСТ Р ИСО 14155-1-2008 Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования
ГОСТ Р ИСО 14155-2-2008 Руководство по проведению клинических испытаний медицинских изделий. Часть 2. Планирование клинических испытаний
ГОСТ Р ИСО 14630-2011 Неактивные хирургические имплантаты. Общие технические требования
ГОСТ Р ИСО 14971-2009 Изделия медицинские. Применение менеджмента риска к медицинским изделиям
ГОСТ Р ИСО 15223-1-2010 Изделия медицинские. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации. Часть 1. Общие требования
ГОСТ Р 51148-98 Изделия медицинские. Требования к образцам и документации, представляемым на токсикологические, санитарно-химические испытания, испытания на стерильность и пирогенность
ГОСТ Р 51609-2000 Изделия медицинские. Классификация в зависимости от потенциального риска применения. Общие требования
ГОСТ Р 52458-2005 (ИСО 11979-7:2001) Имплантаты офтальмологические. Интраокулярные линзы. Часть 7. Клинические испытания
ГОСТ ISO 11137-1-2011 Стерилизация медицинской продукции. Радиационная стерилизация. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий
ГОСТ ISO 11137-2-2011 Стерилизация медицинской продукции. Радиационная стерилизация. Часть 2. Установление стерилизующей дозы
ГОСТ ISO 11607-2011 Упаковка медицинских изделий, подлежащих финишной стерилизации. Общие требования
ГОСТ ISO 13485-2011 Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя "Национальные стандарты" за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 система доставки (delivery system): Герметичный контейнер, в котором поставляется изделие, и дополнительные компоненты для введения данного изделия в глаз.
3.2 динамическая вязкость (dynamic viscosity): Мера внутреннего трения жидкости, равная отношению тангенциального напряжения к градиенту скорости сдвига.
Примечания
1 Термин "Динамическая вязкость" можно также применять для обозначения зависимости от времени, при котором напряжение сдвига и скорость сдвига имеют синусоидальную временную зависимость.
2 Значение динамической вязкости определяют как частное деления касательного напряжения на скорость деформации при синусоидальных условиях.
3 Иногда термин "динамическая вязкость" называют коэффициентом динамической вязкости или просто вязкостью.
4 Динамическая вязкость выражается в паскалях в секунду.
3.3 межфазное натяжение (interfacial tension): Мера избыточной энергии, выделяющейся на границе раздела фаз вследствие дисбаланса молекулярных сил на этой границе.
Примечание - Межфазное натяжение выражается в ньютонах на метр (Н/м).
3.4 кинематическая вязкость (kinematic viscosity): Отношение динамической вязкости жидкости к ее плотности.
Примечание - Кинематическая вязкость выражается в метрах в квадрате в секунду (м/с).
3.5 нетвердые имплантаты (non-solid implants): Тампонада средней оболочки стенки кровеносного сосуда, такая как газы, жидкости или гели.
3.6 поверхностное натяжение (surface tension): Межфазное натяжение в том случае, когда одной из фаз является жидкость, а другой фазой - ее пар.
Примечание - Поверхностное натяжение выражается в ньютонах на метр (Н/м).
3.7 давление пара (vapour pressure): Давление пара жидкой ГЭ, определяющее ее летучесть.
Примечание - Давление паров выражается в миллиметрах ртутного столба (мм рт.ст.) при температуре (35±2)°С.
3.8 глазные эндотампонады (ocular endotamponades): Нетвердые имплантаты, совместимые со средой глаза, применяемые для изменения и/или тампонирования отслоения сетчатки, которые функционируют, в основном, механически.
Примечание - ГЭ применяют либо внутриоперативным введением, либо удалением в конце операции, в случае тяжелых жидкостей, например перфторуглеродов, либо предназначены для нахождения в полости стекловидного тела, пока не будет достигнуто присоединение сетчатки.
3.9 плотность (density): Масса вещества на единицу объема при данной температуре.
3.10 молекулярно-массовое распределение (chain-length distribution): Соотношение числа макромолекул различной молекулярной массы в образце полимера.
Примечание - Молекулярно-массовое распределение является одной из важнейших характеристик синтетических полимеров, определяющей многие их свойства, в частности, механическую прочность.
3.11 исторический (непараллельный) контроль (historical or nonconcurrent control): Способ клинического испытания, при котором результаты сравниваются с данными наблюдения субъекта в прошлом.
4 Общие требования безопасности ГЭ*
________________
* Наименование раздела 4 в бумажном оригинале выделено курсивом. - .
4.1 Требования к применению
4.1.1 Общие требования к применению ГЭ должны соответствовать предусмотренному применению для неактивных хирургических имплантатов по ГОСТ Р ИСО 14630. Дополнительно к ним изготовитель должен описать и документировать функциональные характеристики ГЭ в отношении:
- химического состава и физических свойств;
- предполагаемого хирургического применения;
- условий эксплуатации и максимальной продолжительности контакта с воздействием на глазные ткани.
4.1.2 Область применения ГЭ должна быть определена с учетом действующих нормативных документов, опубликованной клинической и научной литературы, доклинической и клинической оценок и клинических испытаний.
4.2 Требования к конструктивным особенностям и характеристикам
4.2.1 Общие требования
4.2.1.1 Следует применять общие требования к неактивным хирургическим имплантатам по ГОСТ Р ИСО 14630.
4.2.1.2 Все требования, предъявляемые к определениям характеристик, описанные в настоящем подразделе, должны быть соблюдены при испытании стерильного изделия, готового к выпуску.
Любые используемые аналитические методы должны быть подтверждены документально.
Примечание - Указанные требования предназначены для применения при аттестации материалов, но не обязательны к применению в стандартной программе обеспечения/контроля качества.
4.2.2 Требования к химическим и биологическим загрязнениям
Выявление потенциально опасных химических или биологических загрязнителей проводят с помощью анализа риска. Для сырья биологического происхождения эти примеси могут включать в себя белки, нуклеиновые кислоты или другие биологические материалы. Загрязняющие вещества готового продукта, полученные из исходных материалов или в результате производственного процесса, такие как сшивающие агенты и антиоксиданты, которые являются потенциально или системно опасными для тканей глаза, должны быть по возможности определены, и их концентрация в готовой продукции должна сообщаться изготовителем. Загрязняющие вещества должны определяться с помощью стандартных аналитических методов при наличии таковых, и все методы должны быть описаны. Ограничения в отношении выявленных загрязнений должны быть установлены и документированы.
Необходимость проведения испытания для проверки биологического действия загрязнителей в процессе оценки биологической безопасности возникает по результатам анализа рисков.
4.2.3 Требования к химическому описанию
Изготовитель должен представить описание каждого химического компонента в готовой продукции и спецификацию. Если компонент материала происходит из биологических источников, организм, из которого он получается, должен быть указан вместе с его источником. Для синтетических полимеров все повторяющиеся подзвенья, составляющие полимер, должны быть идентифицированы в химическом отношении, все связи между ними должны быть описаны. Также должны быть описаны перекрестные связи. Остаточные мономеры и реакции побочных продуктов должны быть количественно сосчитаны и определены, если это возможно.
4.2.4 Требования к концентрации компонентов
Концентрация каждого компонента материала в готовом изделии должна быть указана изготовителем. Используемые физические или химические методы их определения должны быть указаны.
4.2.5 Требования к плотности
Плотность жидкой формы ГЭ должна быть указана изготовителем в килограммах на кубический метр (кг/м).
4.2.6 Требования к газообразному расширению
Для газообразных форм ГЭ изготовителем должно быть указано внутриглазное газообразное расширение при температуре (35±2)°С и представлена его зависимость от атмосферного давления.
4.2.7 Требования к поверхностному натяжению
Изготовитель должен указать, в случае применимости, поверхностное натяжение в ньютонах на метр (Н/м) при температуре (35±2)°С.
4.2.8 Требования к кинематической вязкости
Изготовитель должен указать, в случае применимости, кинематическую вязкость в миллиметрах в квадрате в секунду (мм/с).
4.2.9 Требования к молекулярно-массовому распределению
Соотношение числа молекул и средние значения молекулярной массы должны быть указаны изготовителем, если ГЭ представляет собой полимер.
Изготовитель должен проводить и сообщать о необходимых дополнительных испытаниях, чтобы обеспечить адекватное описание молекулярно-массового распределения компонентов в готовом изделии. При наличии стандартных методов они должны быть использованы и указаны.
4.2.10 Требования к твердым частицам
При проведении оценки риска необходимо определить потенциальную возможность загрязнения или образования твердых частиц в изделии в процессе изготовления, в условиях, ожидаемых при транспортировании и хранении, а также при предполагаемом использовании изделия.
Изготовитель должен охарактеризовать и установить ограничения на виды, диапазон размеров и число частиц, присутствующих в готовом изделии, используемом в клинических испытаниях. Должны быть установлены предельные значения по каждому виду частиц, подтвержденные в ходе клинических испытаний и подтвержденные соответствующим обоснованием.
4.2.11 Требования к показателю преломления
Показатель преломления ГЭ должен быть измерен с помощью рефрактометра при температуре (35±2)°С и длине волны (546±10) нм.
4.2.12 Требования к спектральному коэффициенту пропускания
Коэффициент пропускания готового ГЭ должен быть измерен в диапазоне от 300 до 1100 нм с помощью спектрофотометра. Результаты должны быть представлены в графической форме: коэффициент пропускания, %, в зависимости от длины волны, нм.
4.2.13 Требования к давлению пара
Давление пара должно быть указано изготовителем, в случае применимости, в миллиметрах ртутного столба (мм рт.ст.) при температуре (35±2)°С.
5 Требования к оценке свойств готового изделия*
________________
* Наименование раздела 5 в бумажном оригинале выделено курсивом. - .
5.1 Общие требования
5.1.1 Требования к оценке ГЭ должны соответствовать требованиям безопасности, приведенным в ГОСТ Р ИСО 14971 для неактивных хирургических имплантатов, общим требованиям ГОСТ Р 51609 и системным требованиям по ГОСТ ISO 13485. Результаты анализа рисков должны определить испытания, необходимые для оценки биологической безопасности ГЭ.
При оценке риска должно быть принято во внимание следующее:
a) вид изделия и продолжительность внутриглазного контакта;
b) потенциальное взаимодействие ГЭ с другими материалами, которые могут быть использованы в офтальмологической операции;
c) для внутриглазных газов любые изменения состава примеси.
Примечание - Изменение состава примесей может происходить вследствие изменения концентрации химических веществ, связанных с различием в давлении паров, когда резервуар будет исчерпан. Требования к оценкам неактивных имплантатов, указанные в ГОСТ Р ИСО 14630, также должны быть применены.
5.1.2 Требования к образцам и документации, представляемым на токсикологические, санитарно-химические испытания, испытания на стерильность и пирогенность, должны соответствовать ГОСТ Р 51148.
5.2 Требования к оценке биологической безопасности
5.2.1 Общие требования
Для всех ГЭ должны быть соблюдены требования, предъявляемые к оценке биологической безопасности по ГОСТ Р ИСО 10993-1, ГОСТ Р ИСО 10993-3, ГОСТ Р ИСО 10993-5, ГОСТ Р ИСО 10993-6, ГОСТ Р ИСО 10993-10, и требования, изложенные в настоящем подразделе.
Примечание - На основании типичного клинического применения в задней камере глаза ГЭ классифицируются как "имплантируемые изделия; ткань/кость". Испытания изделий данной и других категорий, представленные в таблице 1 ГОСТ Р ИСО 10993-1, даны только для руководства и не содержат максимальных или минимальных требований к испытаниям.
5.2.2 Испытание на бактериальные эндотоксины
ГЭ должны быть оценены на наличие бактериальных эндотоксинов с помощью LAL-теста (на культуре Limulus Amebocyte Lysate) в соответствии с методикой, описанной в [1], или эквивалентной аттестованной методикой испытания. Изделие, в котором предел бактериальных эндотоксинов более 0,5 единиц эндотоксинов (ЕЭ) на миллилитр, считают не прошедшим испытание.
5.2.3 Испытание имплантацией внутрь глаза
Испытания на внутриглазное раздражение, воспаление, внутриглазное давление (ВГД) и другие местные явления следует проводить на подходящей модели животного в соответствии с требованиями ГОСТ Р ИСО 10993-2. Должны быть применены частные требования к испытаниям имплантацией внутрь глаза, указанные в приложении А.
При испытании конструктивных особенностей и характеристик ГЭ должны быть оценены дооперационное и послеоперационное внутриглазные раздражения, воспаление, ВГД и другие локальные явления глазной хирургии путем сравнения с контрольной ГЭ, одобренной и прошедшей клинические испытания. Испытуемая ГЭ должна моделировать предполагаемое ее применение с учетом различия между глазами человека и моделью животного.
Послеоперационные явления должны отслеживаться и документироваться с интервалом, соответствующим длительности предполагаемого использования ГЭ.
Если испытуемая ГЭ вызывает большую реакцию глаза или воспаление, чем ГЭ, используемая как контрольная, необходимо провести оценку соотношения риск/польза.
Примечание - Возможно объединить испытания на биологическую безопасность, тем самым уменьшая число животных, необходимых для испытания. Два испытания могут быть проведены с одним животным при условии, что оно не подвергалось неоправданной боли и страданию.
5.3 Требования к клиническим испытаниям
5.3.1 Общие требования
Необходимость клинического испытания ГЭ должна быть установлена в ходе доклинической оценки и оценки риска. Должны быть применены общие требования к клиническим испытаниям по ГОСТ Р ИСО 14155-1 и ГОСТ Р ИСО 14155-2 и требования, приведенные в приложении В.
6 Требования* к стерилизации
________________
* Слово "Требования" в наименовании раздела 6 в бумажном оригинале выделено курсивом. - .
6.1 Изделие должно быть стерилизовано. Следует применять требования к стерилизации, указанные в ГОСТ Р ИСО 14630 для неактивных хирургических имплантатов.
6.2 При стерилизации не следует использовать этиленоксид, если нет документального обоснования для его применения.
В случае применения этиленоксида при использовании в компонентах ГЭ или при документально обоснованном применении в стерилизации предельные значения содержания этиленоксида и этиленхлоргидрина в ГЭ должны составлять 20 и 100 мкг/г соответственно.
6.3 К ГЭ или ее компонентам, при стерилизации которых используется этиленоксид, должны быть применены требования по ГОСТ Р ИСО 10993-7 и ГОСТ Р ИСО 11135.
К ГЭ или ее компонентам, при стерилизации которых используют облучение, должны быть применены требования по ГОСТ ISO 11137-1 и ГОСТ ISO 11137-2.
К ГЭ или ее компонентам, при стерилизации которых используют влажное тепло, должны быть применены требования по ГОСТ Р ИСО 11134.
К ГЭ или ее компонентам, при стерилизации которых используют сухое тепло, должны быть применены требования по [2].
6.4 Если изделие нельзя стерилизовать, применяют асептическую обработку. К ГЭ, которые не стерилизуют, но подвергают асептической обработке, должны быть применены требования по ГОСТ Р ИСО 13408-1. Соответствие ГОСТ Р ИСО 13408-1 должно быть подтверждено аттестованным испытанием в среде с пределом нормы загрязнения 10.
Примечание - В ГОСТ Р ИСО 13408-1 установлены общие требования и приведено руководство по процессам, программам и методикам аттестации и контроля медицинских изделий, подвергаемых асептической обработке. ГОСТ Р ИСО 13408-1, в частности, применим к обработке физиологических (водных) растворов и, следовательно, имеет отношение к изготовлению ГЭ. Следующие части ГОСТ Р ИСО 13408-1 будут посвящены специализированным процессам, таким как фильтрация и лиофилизация.
7 Требования* к стабильности ГЭ
________________
* Слово "Требования" в наименовании раздела 7 в бумажном оригинале выделено курсивом. - .
7.1 Изготовитель должен определить и установить срок годности ГЭ при хранении в складских помещениях и систему поставки. Определение срока годности при хранении должно быть проведено в реальном масштабе времени или проверено по ускоренной методике при температуре не более 45°С для подтверждения, что основные характеристики по безопасности и эффективные эксплуатационные качества готового изделия и системы поставки находятся в установленных пределах в течение обозначенного срока годности при ожидаемых условиях транспортирования и хранения.
7.2 Параметры, которые необходимо контролировать в течение срока годности, являются факторами, установленными при анализе рисков как критические для безопасного применения изделия.
7.3 Изменения состава изделия, исходных материалов и их поставщиков, условий изготовления, включая процесс стерилизации, тип упаковки или упаковочные материалы, могут оказать неблагоприятное влияние на срок годности изделия при хранении. Установленный срок годности ГЭ при хранении должен быть пересмотрен, если при анализе рисков выявлено изменение технологии, что может повлиять на стабильность изделия.
8 Требования* к целостности и эффективности системы доставки
________________
* Слово "Требования" в наименовании раздела 8 в бумажном оригинале выделено курсивом. - .
8.1 Физико-химическая совместимость ГЭ и системы доставки должна быть оценена и документирована.
9 Требования* к упаковке
________________
* Слово "Требования" в наименовании раздела 9 в бумажном оригинале выделено курсивом. - .
9.1 Защита от повреждения при хранении и транспортировании
Должны быть применены требования к упаковке медицинских изделий, указанные в ГОСТ ISO 11607 и ГОСТ Р ИСО 14630. Требования ГОСТ ISO 11607 применимы также для ГЭ, которые не являются абсолютно стерильными.
9.2 Защита стерильности при транспортировании
ГЭ должны быть упакованы таким образом, чтобы оставаться стерильными в пределах, указанных для условий транспортирования, хранения и обращения. Следует соблюдать требования к стерильной упаковке, указанные в [3].
10 Требования* к информации, предоставляемой изготовителем
________________
* Слово "Требования" в наименовании раздела 10 в бумажном оригинале выделено курсивом. - .
10.1 Следует соблюдать общие требования к информации, предоставляемой изготовителем медицинских изделий, указанные в [4], и частные требования к ГЭ, приведенные в настоящем разделе.
10.2 По применимости допускается использовать символы вместо текста. Если используются символы, должны быть применены требования ГОСТ Р ИСО 15223-1.
10.3 Если изделие подвержено действию факторов окружающей среды, на транспортном контейнере должны быть нанесены четкие предупредительные знаки.
10.4 Вкладыш должен быть вложен в упаковку так, чтобы его можно было взять и прочитать, не повреждая стерильного барьера.
10.5 В случае необходимости должна быть обеспечена реабсорбция и представлены расширенная информация, основанная на результатах клинических испытаний, а также предупреждение о диапазоне климатических условий при авиаперелетах.
10.6 При наличии газов должна быть предусмотрена информация о стерильной фильтрации перед внутриглазной инъекцией.
10.7 Номер партии, срок годности и дату стерилизации допускается не указывать на стерильном барьере, если он прозрачный и необходимую информацию можно прочитать на первичном контейнере, не нарушая его герметичность.
Примечание - Номер партии и дату истечения срока годности допускается указывать на самоклеящейся этикетке.
10.8 Минимальная информация, наносимая на упаковочный контейнер, вкладыш в упаковку, стерильный барьер и первичный контейнер, приведена в таблице 1.
Таблица 1 - Информация, предоставляемая изготовителем
Информация | Контейнер для хранения | Вкладыш в упаковку | Стерильный барьер | Первичный контейнер |
Наименование изготовителя или уполномоченного представителя | + | + | - | + |
Адрес изготовителя | + | + | - | - |
Торговое наименование изделия | + | + | - | + |
Краткое описание химического состава изделия и объема поставки | + | + | - | - |
Описание соответствующих конструктивных параметров, которые могут повлиять на безопасность и характеристики изделия | - | + | - | - |
Спектральный коэффициент пропускания (график) | - | + | - | - |
Показатель преломления | - | + | - | - |
Условия хранения | + | + | - | - |
Показания к применению | - | + | - | - |
Противопоказания к применению | - | + | - | - |
Инструкция по применению, включая рекомендации по удалению изделия (при необходимости) | - | + | - | - |
Меры предосторожности и предупреждения | - | + | - | - |
Сообщение о том, что содержимое предназначено для разового использования | + | + | + | - |
Описание процесса подготовки изделия и контейнера к минимальному риску микробиологического заражения | + | + | - | - |
Надпись "СТЕРИЛЬНО" и метод(ы) стерилизации изделия и первичного контейнера | + | + | + | - |
Надпись "Не использовать, если стерильный барьер нарушен" | - | + | + | - |
Конечный срок годности | + | - | + | + |
Номер партии после слова "ЛОТ" | + | - | + | + |
Приложение А
(обязательное)
Методика испытания имплантацией внутрь глаза
А.1 Общие требования
А.1.1 При проведении испытания имплантацией внутрь глаза проводят оценку местного действия на живую ткань образца изделия, имплантированного хирургическим способом в месте, соответствующем предусмотренному применению, маршруту и длительности контакта как на микроуровнях, так и макроуровнях. Общие требования к испытаниям на имплантацию должны соответствовать указанным в ГОСТ Р ИСО 10993-6.
А.1.2 В качестве места для имплантации может быть использована стекловидная полость глаза животного, подходящего для испытания. Выбор модели животного должен быть обоснован. Использование соответствующего контроля должно быть включено в испытание.
В соответствии с требованиями ГОСТ Р ИСО 10993-2 испытание на животном должно быть сокращено до оправданного минимума.
А.2 Порядок проведения испытания
А.2.1 Соответствующий объем ГЭ вводят в стекловидную полость глаза с учетом предполагаемого применения.
А.2.2 Имплантацию осуществляют с минимально возможной травмой глаза с тем, чтобы операционная травма глаза не маскировала повреждения, полученные в результате воздействия испытуемого или контрольного материала.
А.2.3 В качестве контроля используют контрольный образец ГЭ, присутствующий на рынке и предназначенный для тех же целей.
Примечание - Предпочтительна двусторонняя имплантация, но допускается и односторонняя имплантация, если того требуют местные предписания.
А.3 Правила обработки результатов испытания
А.3.1 После введения ГЭ воспалительную реакцию постоянно контролируют и классифицируют в соответствующие промежутки времени измерением внутриглазного давления (ВГД), чтобы выявить периодическую гистологическую оценку, общую и микроскопическую оценки, а также оценку глаза, например глазного дна и щели раздражения, эмульгирование, происхождение катаракты, миграцию материала, статус сетчатки и т.д.
А.3.2 Дополнительные параметры и/или оценки добавляют в зависимости от результата анализа рисков и продолжительности исследования имплантации.
А.3.3 Результаты испытаний должны быть документально оформлены и сообщены согласно требованиям ГОСТ Р ИСО 10993-6.
Приложение В
(справочное)
Требования* к клиническому испытанию
________________
* Слово "Требования" в наименовании приложения В в бумажном оригинале выделено курсивом. - .
В.1 Общие требования
В.1.1 Приведенные в настоящем приложении требования распространяются на ГЭ трех типов, используемых в настоящее время: внутриглазные газы, силиконовое масло и перфторуглеродные соединения в виде жидкости.
В.2 План клинического испытания
В.2.1 Общие требования
В.2.1.1 Должно быть проведено и представлено статистическое клиническое испытание в контролируемых условиях. Целью испытания должна быть документально подтвержденная безопасность новой ГЭ во время операции по сравнению с аналогичной контрольной ГЭ. Анализ риска должен определить исходную гипотезу, а для расчета необходимого числа субъектов в каждой контрольной группе должны быть использованы стандартные биостатистические формулы. Используют рандомизированный (параллельный) или исторический (непараллельный) контроль.
В.2.1.2 В качестве контрольной ГЭ должна быть применена документированная ГЭ, широко реализуемая в течение последних пяти лет, предназначенная и утвержденная для того же применения. Число не доведенных до конца субъектов в каждой контрольной группе должно составлять не более 10% общего числа субъектов, вовлеченных в исследование. Пример расчета числа субъектов для клинических испытаний ВГД приведен в В.3 настоящего приложения. Каждый клинический испытатель сотрудничал более чем с 20 субъектами или более 25% общего числа субъектов, принимающих участие в испытании.
В.2.1.3 Если изготовитель задает дополнительные требования, например, в отношении послеоперационных воздействий ГЭ, то в план клинических испытаний должны быть включены эти требования, а также представлен объем выборки субъектов для участия в испытании этих требований.
В.2.1.4 Длительность использования ГЭ и объем, применяемый для каждого субъекта, должны быть подтверждены документально.
В.2.1.5 Серьезные неблагоприятные операционные и послеоперационные исходы, а также вредные воздействия ГЭ должны быть документально подтверждены и занесены в окончательный отчет в соответствии с требованиями ГОСТ Р 52458.
В.2.2 Требования к клиническим переменным
В.2.2.1 В случае применения исторического контроля методы оценки переменных параметров ГЭ должны соответствовать методам, которые использованы в нем.
В.2.2.2 В процессе исторических испытаний должны быть оценены следующие переменные параметры глаза:
a) наличие или отсутствие хрусталика; в случае наличия хрусталика - оценка его ясности;
b) острота зрения;
c) состояние глаза (кератофакия, афакия, псевдофакия, катаракта).
В.2.2.3 В процессе клинических испытаний должны быть оценены следующие переменные параметры в зависимости от применяемого типа ГЭ:
a) внутриглазные газы (менее 30 сут):
1) ВГД;
2) нарушения роговицы;
3) субретинальная жидкость (газ);
b) силиконовое масло (более 30 сут):
1) ВГД;
2) нарушения роговицы;
3) эмульгирование;
c) перфторуглеродные жидкости (менее одного дня):
1) миграция в субретинальном пространстве;
2) сохраненные перфторуглеродные жидкости.
В.2.2.4 Дополнительные переменные, определяемые оценкой рисков, также должны быть оценены и подтверждены документально.
В.2.3 Послеоперационные оценки
В.2.3.1 Послеоперационные оценки для всех типов ГЭ проводят в следующие моменты времени: 1 день ±4 ч; 1 неделя ±2 дня; 1 месяц ±7 дней; 3 месяца ±2 недели; 6 месяцев ±2 недели.
В.2.3.2 Дополнительный послеоперационный период наблюдения (12±1) мес применяют для продукта, оставшегося в глазе более 30 суток.
В.3 Число субъектов для клинических испытаний
В.3.1 Расчет размера выборки субъектов для проведения клинических испытаний основан на аномалии внутриглазного давления в качестве основного критерия. Аномальное внутриглазное давление является основным вопросом безопасности ГЭ, используемых в настоящее время. Клинические испытания предназначены для того, чтобы показать, что испытуемая ГЭ не хуже контрольного в отношении встречаемости пиков внутриглазного давления ниже 5 мм рт.ст. и выше 25 мм рт.ст. в любое время испытания.
В.3.2 За нулевую гипотезу Н принимают то обстоятельство, когда показатель встречаемости пиков с аномальным внутриглазным давлением
за вычетом показателя контрольной встречаемости пиков внутриглазного давления
превышает минимально обнаруживаемую разность между двумя показателями
.
Альтернативной гипотезой Н принимают то обстоятельство, когда показатель встречаемости пиков с аномальным внутриглазным давлением
за вычетом показателя контрольной встречаемости пиков внутриглазного давления
меньше или равен минимально обнаруживаемой разности между двумя показателями
.
Таким образом:
Н:
![]() ,
,
Н:
![]() .
.
В.3.3 Минимальное число оцениваемых субъектов N, необходимых для каждой испытуемой группы , определяют по формуле
![]() ,
,
где - стандартный нормальный квантиль для уровня доверия;
- стандартный нормальный квантиль для множества (охват вероятности).
В.3.4 При контрольной встречаемости =0,2, минимально обнаруживаемой разности
=0,11, показателей множества (
)=0,80 и
=0,10, необходимое число субъектов в каждой исследуемой группе составляет
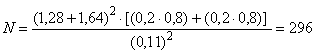 .
.
Приложение ДА
(справочное)
Сопоставление структуры настоящего стандарта со структурой примененного международного стандарта
ДА.1 Сопоставление структуры настоящего стандарта со структурой примененного в нем международного стандарта представлено в таблице ДА.1. Указанное в таблице изменение структуры национального стандарта относительно структуры примененного международного стандарта обусловлено приведением его в соответствие с требованиями ГОСТ Р 1.5.
Таблица ДА.1
Структура международного стандарта ИСО 16672:2003 | Структура настоящего стандарта |
1 Область применения | 1 Область применения |
2 Нормативные ссылки | 2 Нормативные ссылки |
3 Термины и определения | 3 Термины и определения |
4 Область производительности | 4 Общие требования безопасности ГЭ |
4.1 Требования к применению | |
5 Дизайн атрибутов | 4.2 Требования к конструктивным особенностям и характеристикам |
5.1 Общие требования | 4.2.1 Общие требования |
5.2 Химические и биологические загрязнения | 4.2.2 Требования к химическим и биологическим загрязнениям |
5.3 Химическое описание | 4.2.3 Требования к химическому описанию |
5.4 Концентрация компонентов | 4.2.4 Требования к концентрации компонентов |
5.5 Плотность | 4.2.5 Требования к плотности |
5.6 Газообразное расширение | 4.2.6 Требования к газообразному расширению |
5.7 Поверхностное натяжение | 4.2.7 Требования к поверхностному натяжению |
5.8 Кинематическая вязкость | 4.2.8 Требования к кинематической вязкости |
5.9 Молекулярная масса распределения | 4.2.9 Требования к молекулярно-массовому распределению |
5.10 Твердые частицы | 4.2.10 Требования к твердым частицам |
5.11 Показатель преломления | 4.2.11 Требования к показателю преломления |
5.12 Спектральный коэффициент прозрачности | 4.2.12 Требования к спектральному коэффициенту пропускания |
5.13 Поверхностное натяжение | Учтено в 4.2.7 |
5.14 Давление паров | 4.2.13 Требования к давлению пара |
6 Оценка дизайна | 5 Требования к оценке свойств готового изделия |
6.1 Общие положения | 5.1 Общие требования |
6.2 Оценка биологической безопасности | 5.2 Требования к оценке биологической безопасности |
6.2.1 Общие положения | 5.2.1 Общие требования |
6.2.2 Тест на бактериальные эндотоксины | 5.2.2 Испытание на бактериальные эндотоксины |
6.2.3 Тест интраокулярной имплантации | 5.2.3 Испытание имплантацией внутрь глаза |
6.2.4 Окись этилена | Учтено в 6.2 |
6.3 Клинические испытания | 5.3 Требования к клиническим испытаниям |
7 Стерилизация | 6 Требования к стерилизации |
8 Стабильность продукции | 7 Требования к стабильности ГЭ |
9 Целостность и характеристика системы доставки | 8 Требования к целостности и эффективности системы доставки |
10 Упаковка | 9 Требования к упаковке |
11 Информация, предоставляемая изготовителем | 10 Требования к информации, предоставляемой изготовителем |
Приложение А (обязательное) Тест интраокулярной имплантацией | Приложение А (обязательное) Методика испытания имплантацией внутрь глаза |
Приложение В (справочное) Клиническое исследование | Приложение В (справочное) Требования к клиническому испытанию |
- | Приложение ДА (справочное) Сопоставление структуры настоящего стандарта со структурой примененного международного стандарта |
- | Приложение ДБ (справочное) Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации (и действующим в этом качестве межгосударственным стандартам), использованным в настоящем стандарте в качестве нормативных ссылок |
Библиография | Библиография |
Приложение ДБ
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации (и действующим в этом качестве межгосударственным стандартам), использованным в настоящем стандарте в качестве нормативных ссылок
ДБ.1 Сравнение ссылочных международных (региональных) стандартов с национальными стандартами Российской Федерации, использованными в настоящем стандарте в качестве нормативных ссылок, приведено в таблице ДБ.1.
Таблица ДБ.1
Обозначение ссылочного национального, межгосударственного стандарта | Степень соответствия | Обозначение и наименование ссылочного международного стандарта |
IDТ | ИСО 10993-1:1997 "Оценка биологического действия медицинских изделий. Часть 1. Оценка и испытание" | |
IDТ | ИСО 10993-2:1992 "Биологическая оценка медицинских средств. Часть 2. Требования по благополучию животных" | |
- | - | |
ГОСТ Р ИСО 10993-5-1999* | - | - |
________________ * Вероятно, ошибка оригинала. Следует читать: ГОСТ Р ИСО 10993.5-99. - . | ||
ГОСТ Р ИСО 10993-6-1999* | IDТ | ИСО 10993-6:1994 "Оценка биологического действия медицинских изделий. Часть 6. Испытания на местное действие после имплантации" |
________________ * Вероятно, ошибка оригинала. Следует читать: ГОСТ Р ИСО 10993.6-99. - . | ||
ГОСТ Р ИСО 10993-7-1999* | - | - |
________________ * Вероятно, ошибка оригинала. Следует читать: ГОСТ Р ИСО 10993.7-99. - . | ||
ГОСТ Р ИСО 10993-10-1999* | - | - |
________________ * Вероятно, ошибка оригинала. Следует читать: ГОСТ Р ИСО 10993.10-99. - . | ||
- | - | |
- | - | |
- | - | |
- | - | |
IDТ | ИСО 11607-1:2006 "Упаковка медицинских изделий, подлежащих финишной стерилизации. Общие требования" | |
ГОСТ Р ИСО 13408-1-2011 | IDТ | ИСО 13408-1:1998 "Асептическое производство медицинской продукции. Часть 1. Общие требования" |
- | - | |
IDТ | ИСО 14155-1:2003 "Руководство по проведению клинических испытаний медицинских изделий. Часть 1. Общие требования" | |
IDТ | ИСО 14155-2:2003 "Руководство по проведению клинических испытаний медицинских изделий. Часть 2. Планирование клинических испытаний" | |
IDТ | ИСО 14630:1997 "Неактивные хирургические имплантаты. Общие требования" | |
IDТ | ИСО 14971-1:1998 "Медицинские изделия. Менеджмент риска. Часть 1. Применение анализа рисков" | |
IDТ | ИСО 15223:2000 "Медицинские изделия. Символы, применяемые при маркировании на медицинских изделиях, этикетках и сопроводительной документации" | |
- | - | |
- | - | |
NEQ | ИСО 11979-7:2001 "Имплантаты офтальмологические. Интраокулярные линзы. Часть 7. Клинические испытания" | |
Примечание - В настоящем стандарте использованы следующие условные обозначения степени соответствия стандартов: - IDТ - идентичные стандарты; - NEQ - неэквивалентные стандарты. | ||
Библиография
[1] | USP 24 <85> | Фармакопея Соединенных Штатов, 24-е издание, <85> Определение бактериальных эндотоксинов |
(USP 24 <85>) | (United States Pharmacopoeia, 24th revision, <85> Bacterial endotoxins test) | |
[2] | ANSI/AAMI ST 50:1995 | Стерилизаторы сухого тепла (нагретый воздух) |
[3] | EH 868-1:1997 | Упаковочные материалы и системы для медицинских изделий, подлежащих стерилизации. Часть 1. Общие требования и методы испытания |
(EN 868-1:1997) | (Packaging materials and systems for medical devices which are to be sterilized - Part 1: General requirements and test methods) | |
[4] | EH 1041:1998 | Информация, предоставляемая изготовителем медицинской продукции |
(EN 1041:1998) | (Information supplied by the manufacturer with medical devices) |
УДК 681.735:006.354 | ОКС 11.040.40 | Р22 | ОКП 94 3800 |
Ключевые слова: офтальмологические имплантаты, эндотампонады глазные, требования безопасности, характеристики, оценка свойств, клиническая оценка, стерилизация, упаковка, информация, методы оценки | |||
Электронный текст документа
и сверен по:
, 2015
