

ГОСУДАРСТВЕННЫЕ СТАНДАРТЫ СОЮЗА ССР
БЕТОН И ЖЕЛЕЗОБЕТОННЫЕ ИЗДЕЛИЯ
МЕТОДЫ ИСПЫТАНИЙ МАТЕРИАЛОВ
ЧАСТЬ 2
Издание официальное
Москва
ИЗДАТЕЛЬСТВО СТАНДАРТОВ
1 985
ГОСУДАРСТВЕННЫЕ СТАНДАРТЫ СОЮЗА ССР
БЕТОН И ЖЕЛЕЗОБЕТОННЫЕ ИЗДЕЛИЯ
МЕТОДЫ ИСПЫТАНИИ МАТЕРИАЛОВ
ЧАСТЬ 2
Издание официальное
МОСКВА- 1 985
ОТ ИЗДАТЕЛЬСТВА
Сборник «Бетон и железобетонные изделия. Методы йены* таний материалов» часть 2 содержит стандарты, утвержденные до 1 января 1985 г.
В стандарты внесены все изменения, принятые до указанного срока. Около номера стандарта, в который внесено изменение, стоит знак *.
Текущая информация о вновь утвержденных и пересмотренных стандартах, а также о принятых к ним изменениях публикуется в выпускаемом ежемесячно информационном указателе «Государственные стандарты СССР».
30209
085(02)—85
€) Издательство стандартов, 1985
Группа Ж19
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
ГОСТ 5382-73
Взамен
ГОСТ 5382—55
ЦЕМЕНТЫ
Методы химического анализа
Cements. Methods of chemical analysis
Постановлением Государственного комитета Совета Министров СССР по делам строительства от 19 апреля 1973 г. N9 59 срок введения установлен
с 01,01.74
Несоблюдение стандарта преследуется по закону
Настоящий стандарт распространяется на цементы, приготовленные на основе портландцементного клинкера, и устанавливает методы химического анализа.
Химический анализ глиноземистых и гипсоглиноземистых цементов производится по ГОСТ 9552—76.
Методы анализа, изложенные в разд. 2 и 3, являются равноценными по точности получаемых результатов.
Методы химического анализа цементов, изложенные в настоящем стандарте, могут применяться также для анализа шлама, клинкера и шлака. Анализ клинкера и основного шлака производят по методам, изложенным для портландцемента, шлама и кислого шлака — по методам, изложенным для пуццоланового портландцемента.
1. ОБЩИЕ УКАЗАНИЯ
1.1. Средняя проба цемента, шлама или клинкера для химического анализа массой около 200 г должна быть доставлена в лабораторию в стеклянной банке с плотно закрытой пробкой. Пробу квартованием сокращают до 25 г (клинкер предварительно грубо измельчают в стальной ступке), после чего удаляют при помощи магнита металлические частицы, попавшие в пробу цемента. Затем вторичным квартованием отбирают для анализа среднюю аналитическую пробу массой около 10 г, которую окончательно растирают в агатовой, яшмовой или корундовой ступке до прохождения через сетку № 008 по ГОСТ 3584—73, и сохраняют в стаканчике с притертой крышкой.
Издание официальное
Перепечатка воспрещена
Перед взятием навески проба должна быть высушена до постоянной массы при температуре 105—110° и тщательно перемешана. Все определения, за исключением определения содержания влаги, производят из навесок высушенной пробы, именуемых в дальнейшем навеска.
1.2. Взвешивание должно производиться на аналитических весах с точностью до ±0,0002 г.
1.3. При анализе должны применяться только проверенные на чистоту стандартные реактивы квалификации х. ч. и ч. д. а., дистиллированная вода по ГОСТ 6709—72 и беззольные фильтры. Проверяют калибровку применяемой мерной посуды.
1.4. Подсчет результатов анализа производят с точностью до 0,01%. В результаты анализа должна быть введена поправка на загрязнение реактивов, воды и на зольность фильтров.
Величины поправок находят контрольным (холостым) определением и анализом стандартного образца.
1.5. Под концентрацией растворов в процентах имеется в виду содержание граммов вещества в 100 мл раствора.
1.6. Концентрацию разбавленных кислот в ряде случаев выражают как отношение объема кислоты к объему воды. Например, раствор соляной кислоты 1:3 означает, что один объем соляной кислоты плотностью 1,19 разбавлен тремя объемами воды.
1.7. Правильность установки титров стандартных растворов и точность выполнения всех определений контролируют анализом стандартных образцов.
1.8. Отклонения в результатах параллельных определений не должны превышать следующих величин в %.
Потеря при прокаливании
Двуокись кремния
Окись алюминия
Окись железа
Окись кальция
Окись магния
Серный ангидрид
2. МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА
2.1. Определение содержания гигроскопической влаги
В предварительно высушенный и взвешенный стаканчик с притертой крышкой отвешивают около 1 г воздушно-сухой пробы тон-корастертого цемента, помещают стаканчик в сушильный шкаф и сушат пробу при температуре 105—110° С. По истечении 2 ч стаканчик вынимают из шкафа щипцами с каучуковыми наконечниками, закрывают неплотно крышкой и охлаждают в эксикаторе до комнатной температуры. После этого стаканчик взвешивают, пред--
варительно плотно закрыв его крышкой. Для проверки постоянства массы производят повторное высушивание навески при той же температуре в течение 30 мин. Содержание гигроскопической влаги (Н2О) в процентах вычисляют по формуле
Н2О =
лу 100 Мо
где Mi — разность в массе стаканчика с навеской до и после высушивания, г;
А10— навеска пробы воздушно-сухого цемента, г.
Примечание. Если полный анализ цемента выполняется из воздушно-сухой извести Go, то сухую навеску цемента вычисляют по формуле
М = ЛЬ (100- Н2О)
100
2.2. Определение содержания потери при прокаливании
В предварительно прокаленный и взвешенный платиновый или фарфоровый тигель отвешивают около 1 г цемента, высушенного при температуре 105—110° С, и постепенно нагревают его на пламени газовой горелки или в муфельной печи до температуры 950—1000° С, выдерживают при этой температуре в течение 30 мин, а затем охлаждают в эксикаторе и взвешивают. Прокаливание повторяют при той же температуре по 10 мин до получения постоянной массы.
Потерю при прокаливании шлакопортландцемента и шлака определяют прокаливанием навески около 1 г в муфельной печи при температуре 900—950° С в течение 1—2 мин, навеску охлаждают в эксикаторе и взвешивают. Прокаливание повторяют в течение 2—3 мин при той же температуре до прекращения уменьшения в массе прокаливаемой пробы.
Потерю при прокаливании (п.п.п.) в процентах вычисляют по формуле где Ah — разность в массе тигля до и после прокаливания, г;
М — навеска цемента, г.
2.3. Определение содержания нерастворимого остатка
Содержание нерастворимого остатка определяют в клинкере либо в цементе, не содержащем минеральных добавок, кроме гипса.
2.3.1. Применяемые реактивы и растворы
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 1 :9.
Натрий углекислый безводный по ГОСТ 83—79, 5%-ный раствор.
Натрия гидрат окиси по ГОСТ 4328—77, 1%-ный раствор.
Аммоний азотнокислый по ГОСТ 22867—77 или аммоний хлористый по ГОСТ 3773—72, 2%-ные растворы.
Метиловый красный индикатор по ГОСТ 5853—51, 0,2%-ный раствор на этиловом синтетическом спирте по ГОСТ 11547—80 или гидролизном техническом этиловом спирте по ГОСТ 17299—78.
Серебро азотнокислое по ГОСТ 1277—75, 1'%-ный раствор.
2.3.2. Проведение анализа
Навеску 1 г цемента помещают в стакан вместимостью 150 мл„ прибавляют при помешивании 25 мл воды и 5 мл соляной кислоты. Растирают навеску плоским концом стеклянной палочки и доводят объем раствора водой до 50 мл. Затем стакан накрывают часовым стеклом и помещают на кипящую водяную баню. По истечении 15 мин жидкость фильтруют через неплотный беззольный фильтр и промывают остаток горячей (60—70°С) водой до исчезновения реакции на ион хлора (проба раствором азотнокислого серебра, подкисленного азотной кислотой). Остаток вместе с фильтром переносят в стакан, в котором производилось разложение цемента, и приливают при помешивании 30 мл раствора углекислого натрия, нагретого до температуры 80—90° С. После этого стакан накрывают часовым стеклом, помещают на плитку с асбестовой сеткой и нагревают в течение 15 мин при температуре, близкой к кипению. Жидкость фильтруют через двойной неплотный беззольный фильтр, остаток промывают 5—6 раз горячей водой (60—70°С), затем смачивают 10—12 каплями раствора соляной кислоты и снова промывают горячей водой до исчезновения реакции на ион хлора.
Остаток после отделения солянокислого фильтра может быть обработан вместо углекислого натрия 100 мл горячего раствора гидрата окиси натрия при температуре, близкой к точке кипения в течение 15 мин. Затем раствор нейтрализуют соляной кислотой по метиловому красному (индикатору) и добавляют еще избыток 4—5 капель той же кислоты. Фильтруют и промывают остаток 10—12 раз горячим раствором азотнокислого или хлористого аммония. После этого остаток с фильтром прокаливают в платиновом или фарфоровом тигле при температуре 900—1000° С до постоянной массы.
Содержание нерастворимого остатка (н. о.) в процентах вычисляют по формуле
где Aft — разность в массе тигля с осадком и пустого тигля, г; М — навеска цемента, г.
Примечания:
1 - При содержании в цементе нерастворимого остатка выше 0,4%, а также при анализе барийсодержащего портландцемента необходимо проверять его на чистоту отгонкой с HF по п. 2.4.2. За величину нерастворимого остатка при этом принимают содержание отогнанной S1O2. Если проверка на чистоту нерастворимого остатка обычного цемента не производилась, то полученную величину умножают на коэффициент 0,7.
2. Солянокислый фильтрат после отделения нерастворимого остатка может быть использован для определения в нем серного ангидрида по п. 2.10.
2.4. Определение содержания двуокиси кремния в портландцементах и шлакопортландце-ментах методом коагуляции
Желатина способствует коагуляции кремнекислоты и при нагревании создает условия для быстрого и количественного перевода ее в нерастворимое состояние. При коагуляции в концентрированном растворе соляной кислоты уменьшается соосаждение посторонних ионов и осадок кремнекислоты практически не загрязнен.
2.4.1. Применяемые реактивы и растворы
Кислота серная по ГОСТ 4204—77, плотностью 1,84.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 5:95.
Желатина, 1%-ный раствор, свежеприготовленный; готовят следующим образом: 1 г желатины растворяют в 100 мл воды, нагретой до 70° С.
Кислота фтористоводородная (плавиковая) по ГОСТ 10484—78, 40%-ная. Чистоту фтористоводородной кислоты проверяют выпариванием 7—10 мл ее с 3—4 каплями серной кислоты в платиновом тигле, остаток прокаливают при температуре 950—1000° С и взвешивают. При большом остатке необходимо вводить поправку в результат определения двуокиси кремния.
Серебро азотнокислое по ГОСТ 1277—75, 1%-ный раствор.
Кислота азотная по ГОСТ 4461—77, плотностью 1,4.
2.4.2. Проведение анализа
Навеску 0,5 г цемента помещают в сухой стакан вместимостью 50 мл, накрывают часовым стеклом и осторожно добавляют 10 мл соляной кислоты так, чтобы кислота стекала по стенке стакана. Стакан погружают в нагретую до 60—70° С водяную баню и выдерживают в течение 10 мин, перемешивая время от времени содержимое. Затем прибавляют 10 мл раствора желатины, энергично перемешивают в течение 1 мин, не вынимая стакан из водяной бани, и нагревают еще в течение 10 мин. Раствор охлаждают до 40—50° С и в теплом виде фильтруют через неплотный беззольный фильтр, затем тщательно удаляют приставшие к стенкам стакана частички кремнекислоты и присоединяют их к осадку. Осадок на фильтре промывают 2—3 раза горячим раствором соляной кислоты, а затем 10—12 раз небольшими порциями горячей воды (температурой не выше 70°С), давая полностью стечь каждой порции.
Осадок вместе с фильтром переносят во взвешенный платиновый тигель, озоляют без воспламенения, прокаливают в муфельной печи в течение 20—25 мин при температуре 1050—1100° С, охлаждают и взвешивают. Прокаливание повторяют до получения постоянной массы.
Содержание двуокиси кремния (SiOs) в процентах вычисляют по формуле
- 100
где Mi — привес тигля, г;
М— навеска цемента, г.
Фильтрат сохраняют для определения остальных компонентов.
Полученную кремнекислоту проверяют на чистоту. Для этого осадок в тигле смачивают несколькими каплями воды, прибавляют 3—4 капли серной кислоты и 7—10 мл фтористоводородной кислоты и выпаривают содержимое тигля на песочной бане до прекращения выделения паров серной кислоты. После этого тигель прокаливают при температуре 1050—1100° С в течение 3—5 мин, охлаждают в эксикаторе и взвешивают.
Содержание остатка после отгона (остаток) в процентах вычисляют по формуле
Остаток ~ ———
М
100,
где Л11 — масса тигля с прокаленным остатком после выпаривания с плавиковой кислотой, г;
М2 — масса пустого тигля, г;
М — навеска цемента, г.
При величине остатка более 0,5% определение кремнекислоты необходимо повторить весовым (п. 2.5.2) или фотоколориметри-ческим (п. 3.1) методами.
При анализе цементов, содержащих титан, хром и др., а также для получения результатов повышенной точности (анализы стандартных образцов) помимо проверки на чистоту необходимо определить содержание кремнекислоты в фильтрате после ее отделения.
В этом случае фильтрат после отделения кремнекислоты подкисляют 1—2 мл азотной кислоты, кипятят 2—3 мин для разрушения желатины, затем переводят в мерную колбу вместимостью 250 мл, доводят до метки водой, тщательно перемешивают и определяют содержание кремнекислоты колориметрическим методом по п. 3.1.
Содержание двуокиси кремния (SiCM в процентах вычисляют по формуле
Cs0 _ Mi • 100 cjo
^1ий----Г Ы'Лфидьтр»
где Mi — разность в массе тигля с прокаленным осадком до и после выпаривания с плавиковой кислотой, г;
М — навеска цемента, г;
SiO20H-4bTp — содержание SiO2 в фильтрате в процентах, определенное по п. 3.1.
Отделение гидроокисей и определение содержания остальных составляющих производят из аликвотной части раствора фильтрата от SiO2.
Примечания:
1. При содержании в портландцементах минеральных добавок, нерастворимых в соляной кислоте, а в шлакопортландцементах — кислых шлаков, определение содержания двуокиси кремния производят по методу, указанному в п. 2.5.
2. Определение содержания кремнекислоты в барнйсодержаших портландцементах производят фотоколориметрическим методом по п. 3.1.
3. При величине остатка после отгона кремнекислоты более 0,5% необходимо ее сплавить, растворить в небольшом количестве соляной кислоты и прибавить к основному фильтрату.
2.5. Определение содержания двуокиси кремния в пуццолановых портландцементах методом коагуляции
2.5.1. Применяемые реактивы и растворы
Натрий углекислый безводный по ГОСТ 83—79.
Остальные реактивы — по п. 2.4.
2.5.2. Проведение анализа
Навеску 0,5 г цемента тщательно перемешивают в платиновом тигле примерно с двукратным количеством безводного углекислого натрия. Тигель накрывают крышкой и спекают его содержимое в муфельной печи при температуре 950—1000° С в течение 3—7 мин. После быстрого охлаждения тигля полученный спек растворяют 10—15 мл соляной кислоты, которую приливают небольшими порциями и переносят в стакан вместимостью 100 мл. Тигель ополаскивают этой же кислотой и сохраняют для дальнейшей работы. Стакан накрывают часовым стеклом и погружают в нагретую до 65—70° С водяную баню на 10 мин. После растворения спека прибавляют 10 мл раствора желатины и выполняют все остальные операции выделения и определения кремнекислоты по методу коагуляции желатиной, изложенному в п. 2.4.
Фильтрат сохраняют для определения содержания остальных компонентов.
2.6. Отделение гидроокисей алюминия, железа и т и т а и а
А. Раствором уротропина
Принцип метода. Осаждение гидроокисей основано на гидролизе уротропина в слабокислом растворе с образованием аммиака и формальдегида. Аммиак, медленно выделяющийся при гидролизе уротропина, постепенно увеличивает pH анализируемого раствора* Буферная система аммиак-формальдегид обеспечивает несколько меньшую и благоприятную величину pH раствора (6—6,5) по сравнению с системой аммиак — хлористый аммоний. В растворе при этом также не будет избытка аммонийных солей, затрудняющих определение эквивалентной точки при комплексонометрическом титровании кальция. Применение уротропина (гексаметилентетрамина) вместо аммиака для осаждения гидроокисей алюминия и железа освобождает от их переосаждения вследствие образования плотных осадков гидроокисей, обладающих малой адсорбционной способностью, которые легко и быстро фильтруются, а это существенно сокращает время выполнения анализа.
2.6.1. Применяемые реактивы и растворы
Уротропин- гексаметилентетрамин, 20%-ный водный раствор.
Кислота азотная по ГОСТ 4461—77, плотностью 1,4.
Аммиак водный по ГОСТ 3760—79, 10’%-ный раствор.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 1 :4.
Аммоний азотнокислый по ГОСТ 22867—77, 2%-ный раствор, нейтрализованный аммиаком до пожелтения индикатора метилового красного.
Метиловый красный (индикатор) по ГОСТ 5853—51, 0,2%-ный спиртовой раствор; готовят по п. 2.3 настоящего стандарта.
Серебро азотнокислое по ГОСТ 1277—75, 1%-ный раствор.
2.6.2. Проведение анализа
Фильтрат, после отделения кремнекислоты по п. 2.4 или 2.5, нагревают до кипения, добавляют 7—10 капель азотной кислоты, слабо кипятят раствор еще 1—2 мин, после чего нейтрализуют раствором аммиака до начала покраснения бумажки конго, затем добавляют несколько капель раствора соляной кислоты (1 :4) до перехода цвета бумажки конго в сиреневый (или до появления слабой мути, которую растворяют несколькими каплями соляной кислоты 1:4), приливают 20 мл раствора уротропина и раствор выдерживают в течение 10 мин при температуре около 80° С, избегая его кипения. Осадок фильтруют через неплотный беззольный фильтр, промывают горячим раствором азотнокислого аммония до исчезновения реакции на ион хлора (проба раствором азотнокислого серебра, подкисленного азотной кислотой). Фильтрат с промывными водами переводят в мерную колбу вместимостью 250 мл и сохраняют для определения содержания окисей кальция и магния.
Примечание. Осадок гидроокисей может быть использован для определения содержания окисей железа и алюминия.
Б. Раствором аммиака
2.6.3. Проведение анализа.
Фильтрат после отделения кремнекислоты переносят в стакан вместимостью 300 мл и выпаривают до объема не более 100 мл. Затем прибавляют 7—10 капель азотной кислоты, доводят раствор до кипения и слабо кипятят 1—2 мин. Для того, чтобы присутствующий в аммиаке в небольшом количестве углекислый аммоний не привел к увлечению в осадок некоторого количества углекислого кальция, осаждение гидроокисей выполняют в две стадии.
Сначала к раствору добавляют основное количество аммиака до появления осадка, который затем растворяют несколькими каплями соляной кислоты 1:4; раствор кипятят для удаления углекислоты. При этом выпавший в осадок углекислый кальций растворяется в соляной кислоте.
Затем к раствору добавляют 1—2 капли индикатора метилового красного и окончательно осаждают гидроокиси, прибавляя аммиак по каплям до пожелтения раствора. Окончательное осаждение гидроокисей достигается прибавлением небольшого количества аммиака, которое практически не вызовет осаждения углекислого кальция. Стакан с осадком помещают на несколько минут (до просветления жидкости над осадком) в теплое место, после чего фильтруют через неплотный беззольный фильтр. Осадок промывают 3—4 раза горячим раствором азотнокислого аммония. Затем осадок смывают с фильтра в стакан, в котором проводилось осаждение, прибавляют 30 мл раствора соляной кислоты, разбавляют примерно до 100 мл горячей водой, прибавляют 1—2 капли индикатора метилового красного, вторично осаждают гидроокиси раствором аммиака, фильтруют через тот же фильтр и промывают осадок горячим раствором азотнокислого аммония до исчезновения реакции на ион хлора (проба раствором азотнокислого серебра, подкисленного азотной кислотой). Объединенные фильтраты с промывными водами переводят в мерную колбу вместимостью 250 мл и сохраняют для определения содержания окисей кальция и магния.
2.7. Определение содержания окисей кальция и магния комплексонометрическим методом
Принцип метода. Метод комплексонометрического титрования основан на быстром образовании бесцветных, хорошо растворимых, прочных и незначительно диссоциирующих комплексных соединений трилона Б (комплексона III) с катионами определяемых окислов. Грамм-молекула металла независимо от валентности последнего всегда связывает грамм-молекулу трилона Б.
Для установления конечной точки титрования применяют специальные комплексонометрические индикаторы — металл-индика-торы, образующие с определяемыми ионами окрашенные комплексы менее прочные, чем соответствующие комплексы этих ионов с трилоном Б. В конечной точке титрования при полном связывании в комплекс титруемых ионов раствор окрашивается в цвет, присущий свободному индикатору.
В аликвотной части раствора после отделения гидроокисей алюминия и железа титруют окись кальция трилоном Б при pH выше 12, что обеспечивается введением раствора крепкой калиевой или натриевой щелочи. В качестве индикатора применяют мурексид или флоурексон.
Во второй аликвотной части раствора определяют сумму кальция и магния титрованием раствором трилона Б с индикатором кислотным хром темно-синим или хромогеном черным ЕТ-00.
2.7.1. Применяемые реактивы и растворы
Трилон Б по ГОСТ 10652—73, 0,05 м раствор; готовят следующим образом: 18,6 г трилона Б растворяют в 1 л воды и хорошо перемешивают. Если раствор получился мутным, его фильтруют. Титр раствора устанавливают по кальцию углекислому по ГОСТ 4530—76 с проверенным содержанием СаО или по стандартному образцу известняка 59в. 5 г углекислого кальция или стандартного образца 59в растворяют в 30—40 мл соляной кислоты 1 :3 при нагревании, раствор кипятят 3—5 мин для удаления углекислоты, переводят в мерную колбу вместимостью 1 л, охлаждают, разбавляют водой до метки и хорошо перемешивают. Для установки титра раствора трилона Б спускают из бюретки по 20 мл приготовленного раствора хлористого кальция в три конические колбы вместимостью 250 мл, разбавляют водой до 80—100 мл, приливают из бюретки 10—15 мл устанавливаемого раствора трилона Б, 15 мл раствора гидрата окиси калия (или гидрата окиси натрия), добавляют на кончике шпателя индикатора мурексида, после чего дотитровывают трилоном Б до перехода малиновой окраски раствора в устойчивую фиолетовую. При титровании с индикатором флуорексоном в колбы с анализируемыми растворами вводят такой же избыток раствора трилона Б, добавляют 4—5 капель бромкрезолового пурпурового, 15 мл раствора гидрата окиси калия, на кончике шпателя индикатора флуорексона, после чего продолжают титрование раствором трилона Б до перехода флуоресцирующей малиново-зеленой окраски в устойчивую малиновую. Титрование в этом случае проводят на темном фоне.
Титр раствора трилона Б, выраженный в граммах СаО (7сао), вычисляют по среднеарифметическому результату трех титрований по формуле
т = 20.5. %СаО
СаО V- 100.1000 ’
где % СаО — содержание СаО в углекислом кальции или в стандартном образце 59в, %;
V — объем 0,05 м раствора трилона Б, пошедший на титрование, мл.
Титр раствора трилона Б по MgO устанавливают по сернокислому магнию. Спускают из бюретки 20 мл 0,1 н титрованного раствора сернокислого магния, приготовленного из фиксанала, в три конические колбы вместимостью 250—300 мл. Разбавляют растворы до 100 мл водой, нагревают до 60—70° С, приливают по 15 мл аммиачного буферного раствора и 5—7 капель индикатора кислотного хрома темно-синего или вводят на кончике шпателя сухую смесь индикатора хромогена черного ЕТ-00 (0,04—0,05 г) и титруют 0,05 м раствором трилона Б при интенсивном помешивании до перехода красной окраски соответственно в устойчивую синесиреневую или в синюю с зеленоватым оттенком.
Титр 0,05 м раствора трилона Б, выраженный в граммах MgO (^Mgo), вычисляют по среднеарифметическому результату трех титрований по формуле
« 20.0,002016
1 MgO =----------9
где V — объем 0,05 м раствора трилона Б, пошедший на титрование 20 мл 0,1 н раствора сернокислого магния, мл; 0,002016 — количество окиси магния, соответствующее 1 мл точно 0,05 м раствора трилона Б, г.
Калия гидрат окиси или натрия гидрат окиси по ГОСТ 4328—77, 20%-ные растворы.
Кислота соляная по ГОСТ 3118—77, раствор 1 :3.
Мурексид (индикатор), сухая смесь; готовят следующим образом: 1 г мурексида хорошо перемешивают с 99 г высушенного хлористого натрия безводного по ГОСТ 4233—77 или хлористого калия по ГОСТ 4234—77, помещают в банку с притертой крышкой и хранят в темном месте.
Флуорексон (индикатор), сухая смесь; готовят следующим образом: хорошо перемешивают 1 г флуорексона с 99 г хлористого калия по ГОСТ 4234—77 и сохраняют в банке с крышкой.
Бром крезоловый пурпуровый, 0,1%-ный спиртовой раствор. Маскирующий реагент, 5%-ный водный раствор триэтаноламина, содержащий 0,5% натрия фтористого по ГОСТ 4463—76.
Гидроксиламин солянокислый по ГОСТ 5456—79, 5%-ный раствор.
Аммиачный буферный раствор; готовят следующим образом: растворяют 70 г аммония хлористого по ГОСТ 3773—72 в 200 мл воды, фильтруют, прибавляют 570 мл 25%-ного раствора аммиака по ГОСТ 3760—79, доводят до 1 л водой и хорошо перемешивают; pH этого раствора соответствует примерно 10.
Кислотный хром темно-синий (индикатор), 0.5%-ный водный раствор.
Сухая смесь (индикатора) хромогена черного специального ЕТ-00; готовят следующим образом: перемешивают путем растирания 1 г индикатора с 99 г безводного натрия хлористого по ГОСТ 4233—77 или калия хлористого по ГОСТ 4234—77 и сохраняют в банке с крышкой.
2.7.2. Определение содержания окиси кальция
Из мерной колбы вместимостью 250 мл после отделения гидроокисей отбирают пипеткой 50 мл раствора, переносят его в коническую колбу вместимостью 250 мл, предварительно добавляют 15 мл раствора трилона Б (во избежание выпадения гидрата окиси кальция в щелочном растворе), 20—25 мл раствора гидрата окиси калия (или гидрата окиси натрия), на кончике шпателя индикатора мурексида, после чего дотитровывают раствором трилона Б до перехода малиновой окраски раствора в устойчивую фиолетовую. Титрование с индикатором флуорексоном производят так, как это изложено при установке титра раствора трилона Б.
Содержание окиси кальция (СаО) в процентах вычисляют по формуле
_ V- ТСаО -5 . 100 СаО ----,
м
где V — объем 0,05 м раствора трилона Б, пошедший на титрование, мл;
Тсао — титр 0,05 м раствора трилона Б, выраженный в г СаО;
М— навеска цемента, г;
5 — коэффициент, учитывающий определение СаО в аликвотной части раствора.
Примечания:
1. При анализе шлакопортландцементов и шлаков для предотвращения окисления закиси марганца в щелочном растворе кислородом воздуха перед титрованием кальция приливают 1—3 мл солянокислого гидроксиламина, после чего добавляют все необходимые растворы и титруют трилоном Б.
2. Определение содержания окиси кальция может быть выполнено также из отдельной навески без отделения полуторных окислов после сплавления 0,1 г анализируемого образца с 1 г плавня и последующего растворения сплава в соляной кислоте. Мешающее влияние полуторных окислов устраняют введением (перед добавлением избытка трилона Б) маскирующего реагента в количестве 10 мл.
2.7.3. Определение содержания окиси магния
Из мерной колбы вместимостью 250 мл после отделения гидроокисей по п. 2.6 отбирают пипеткой 50 мл раствора, переносят его в коническую колбу вместимостью 250 мл, разбавляют водой до 100 мл, добавляют 15 мл буферного раствора, 7—8 капель индикатора кислотного хрома темно-синего или на кончике шпателя индикатора хромогена черного ЕТ-00 и титруют сумму окисей кальция и магния 0,05 м раствором трилона Б до перехода красной окраски раствора соответственно в устойчивую сине-сиреневую или в синюю с зеленоватым оттенком.
Содержание окиси магния (MgO) в процентах вычисляют по формуле
-ТМго-5 - 100 м
где Уг — объем 0,05 м раствора трилона Б, пошедший на титрование суммы СаОЧ-MgO, мл;
Vi — объем 0,05 м раствора трилона Б, пошедший на титрование СаО, мл;
TMgo— титр 0,05 м раствора трилона Б, выраженный в г MgO. Значения остальных обозначений такие же, как при определении СаО.
Примечание. Содержание окиси магния может быть также определено в растворе после титрования кальция. Для этого к раствору добавляют соляной кислоты до посинения бумажки конго, для разрушения мурексида и растворения выпавшей гидроокиси магния. Затем приливают 15—20 мл аммиачного буферного раствора, один из индикаторов и титруют магний трилоном Б аналогично вышеизложенному.
Содержание окиси магния (MgO) в процентах вычисляют по формуле
V • T'MgO * 5 ’ I00 м
где V — объем 0,05 м раствора трилона Б, пошедший на титрование, мл.
2.8. Определение содержания закиси железа в шлакопортландцементах
2.8.1. Применяемые реактивы, растворы и аппаратура
Кислота серная по ГОСТ 4204—77, раствор 1 :4.
Калий марганцовокислый по ГОСТ 20490—75, 0,1 н титрованный раствор; готовят из фиксанала.
Углекислый газ; получают в аппарате Киппа действием раствора соляной кислоты на мраморную крошку или известняк или используют углекислый газ из баллонов.
Натрий углекислый безводный по ГОСТ 83—79 плавленый; готовят путем плавления соды в платиновой чашке в муфельной печи при температуре 850—900° С в течение 5—8 мин, после охлаждения разбивают на кусочки.
Клапан Бунзена (черт. 1) изготовляют из толстостенной резиновой трубки. Отрезок такой трубки длиной 5 см с одного конца плотно закрывают резиновой пробкой, иногда заливают резиновым клеем. Другой конец надевают на стеклянную трубку. Лезвием бритвы вдоль резиновой трубки делают прорез (щель) длиной в 1,5—2 см. При проведении анализа коническую колбу вместимостью 250 мл закрывают резиновой пробкой, в которую вставлен стеклянный конец клапана Бунзена.
гост sm-n
Клапан Бунзена
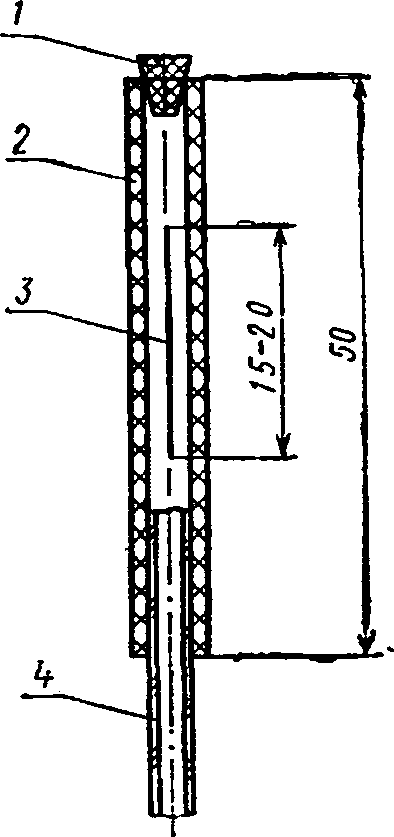
/ — пробка; 2 — резиновая трубка; 3— щель; 4 — стеклянная трубка
Черт. 1
А. Разложение навески в токе углекислого газа
2.8.2. Проведение анализа
В коническую колбу вместимостью 250 мл приливают 100 мл раствора серной кислоты. Колбу закрывают пробкой с двумя отверстиями, в которые вставлены стеклянные трубки, согнутые под прямым углом. Одна из трубок (по ходу газа) доходит до дна колбы, вторая— кончается под пробкой. Длинную трубку присоединяют к аппарату Киппа с углекислым газом. Открыв кран у аппарата, пропускают ток углекислого газа через колбу в течение 3 мин. В это время отвешивают на сухом часовом стекле 1—1,5 г свежеизмель-ченного клинкера или цемента. Приоткрыв пробку, быстро всыпают навеску в колбу, не прекращая тока газа, взвешивают стекло и по разности масс определяют навеску. Содержимое колбы кипятят 15 мин, пропуская все время ток углекислого газа. Затем снимают колбу с плитки и, не прекращая тока углекислоты, охлаждают содержимое колбы, после чего отсоединяют колбу от прибора Киппа, прибавляют 100 мл прокипяченной холодной воды и титруют 0,1 н раствором марганцовокислого калия до розовой окраски, не исчезающей в течение 20—30 с.
Содержание закиси железа (FeO) в процентах вычисляют по формуле
РеО _ V .0,007131 ■ 100 ~ м
где V — объем 0,1 н раствора марганцовокислого калия, пошедший на титрование, мл;
М — навеска цемента, г;
0,007184 — количество закиси железа, соответствующее 1 мл точно 0,1 и раствора марганцовокислого калия, г.
Б. Разложение навески с применением плавленой соды
2.8.3. Проведение анализа
В коническую колбу вместимостью 250 мл вливают 100 мл раствора серной кислоты, всыпают навеску цемента 1—1,5 г, бросают несколько кусочков плавленой соды и быстро закрывают резиновой пробкой с клапаном Бунзена. Содержимое колбы в течение 10—15 мин кипятят на плитке. Затем снимают колбу, охлаждают, прибавляют 100 мл прокипяченной холодной воды; все остальные операции выполняют, как изложено в п. 2.8.2.
Примечание. При наличии в портландцементах и пуццолановых портландцементах закиси железа определение ведут по методам, изложенным выше, а процентное содержание окиси железа вычисляют по формуле для шлакопорт-ландцементов (п. 2.9).
2.9. Определение содержания окисей железа и алюминия трилонометрическим методом
Принцип метода. Содержание окиси железа и алюминия определяют путем последовательного титрования трилона Б при различных значениях pH раствора; железо титруют при pH раствора около 1—2 с сульфосалицилатом натрия в качестве индикатора. В этих условиях присутствие SiO2, А12О3, СаО и MgO не мешает определению Fe2O3, а содержание окиси алюминия определяют обратным титрованием предварительно введенного избытка трилона Б раствором соли трехвалентного железа при pH 4,8—5,0. При повышении pH до указанной величины образовавшийся ранее три-лонат железа не разрушается и не препятствует определению алюминия.
2.9.1. Применяемые реактивы и растворы
Плавень — смесь натрия углекислого безводного по ГОСТ 83—79 и натрия тетраборнокислого (буры) по ГОСТ 4199—76 (безводной) в отношении по массе 2: 1. В плавень рекомендуется добавлять 0,5 г селитры на 100 г плавня для полного окисления железа и сульфидной серы.
Аммиак водный по ГОСТ 3760—79, 10%-ный раствор.
Натрия гидрат окиси по ГОСТ 4328—77 или калия гидрат окиси, 20%-ные растворы.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 1 :3.
Кислота азотная по ГОСТ 4461—77, плотностью 1,4.
Сульфосалициловый индикатор; готовят следующим образом: навеску 10 г сульфосалициловой кислоты по ГОСТ 4478—78 растворяют в 50 мл воды, нейтрализуют 20%-ным раствором щелочи до изменения синей окраски бумажки конго красной на сиреневую и разбавляют водой до 100 мл.
Ацетатный буферный раствор; готовят следующим образом: 165 г плавленого натрия уксуснокислого по ГОСТ 199—78 или 270 г трехводной соли растворяют в 300—400 мл воды, отфильтровывают, добавляют 60 мл ледяной или 75 мл 80%-ной кислоты уксусной по ГОСТ 61—75, разбавляют водой до 1 л и хорошо перемешивают; pH этого раствора соответствует примерно 5.
Титрованный раствор соли трехвалентного железа; готовят следующим образом: растворяют 13,5 г хлорного железа по ГОСТ 4147—74 в 300 мл воды. Раствор отфильтровывают в мерную колбу вместимостью 1 л, добавляют 8—10 мл соляной кислоты, разбавляют водой до метки и хорошо перемешивают. Титр этого раствора устанавливают весовым методом в параллельных пробах — осаждением 10%-ным раствором аммиака из 25 мл раствора в виде гидроокиси с последующим промыванием горячим раствором азотнокислого аммония до исчезновения реакции на ион хлора и прокаливанием осадка при температуре 950—1000° С в течение 20—25 мин.
Вместо хлорного железа можно использовать железоаммонийные квасцы по ГОСТ 4205—77. В этом случае 24,1 г FeN4 (SO4)2X Х12Н2О растворяют аналогичным образом.
Титр раствора хлорного железа, выраженный в граммах F2O3 (а), вычисляют по формуле где М — масса прокаленного осадка Fe2O3, г.
Титрованный раствор соли алюминия; готовят следующим образом: растворяют 16 г квасцов алюмокалиевых KAI (SO4)2*12H2O по ГОСТ 4329—77 в 300 мл воды. Раствор отфильтровывают в мерную колбу вместимостью 1 л, добавляют 9 мл соляной кислоты, разбавляют водой до метки и хорошо перемешивают. Титр этого раствора устанавливают также весовым методом в параллельных пробах после двукратного осаждения гидроокиси алюминия аммиаком по индикатору метиловому красному из 25 мл раствора и последующего прокаливания осадка при температуре 1100—1200°С.
Титр раствора алюмокалиевых квасцов, выраженный в граммах А12О3 (б), вычисляют по формуле
где М — масса прокаленного осадка А12О3, г.
Трилон Б по ГОСТ 10652—73, 0,05 м раствор; готовят по п. 2.7.1.
Титр этого раствора для определения окиси железа устанавливают следующим образом. Спускают из бюретки по 20 мл раствора хлорного железа в три конические колбы вместимостью 250 мл, разбавляют водой до 100 мл, нагревают примерно до 50°С, добавляют по 6—7 капель сульфосалицилового индикатора и титруют раствором трилона Б до исчезновения фиолетовой окраски сульфосалицилата железа.
Титр раствора трилона Б, выраженный в граммах Fe2O3 (Т Fe,o3), вычисляют по среднему арифметическому результату трех титрований по формуле
лр 20. а
* Ге8О3= —— ,
где а — титр раствора хлорного железа, выраженный в г Fe2O3;
V — объем 0,05 м раствора трилона Б, пошедший на титрование, мл.
Устанавливают соотношение между концентрациями растворов трилона Б и хлорного железа. Для этого спускают из бюретки в три конические колбы вместимостью 250 мл по 10 мл раствора трилона Б, разбавляют водой до 100 мл, приливают 10 мл ацетатного буферного раствора, 6—7 капель сульфосалицилового индикатора и титруют раствором хлорного железа до появления золотистооранжевой окраски, не исчезающей в течение 1 мин.
Коэффициент (К), выражающий соотношение между концентрациями растворов трилона Б и хлорного железа, вычисляют как среднее арифметическое из трех определений по формуле
где V — объем раствора хлорного железа, пошедший на титрование 10 мл 0,05 м раствора трилона Б, мл.
Титр раствора трилона Б для определения окиси алюминия устанавливают следующим образом. Спускают из бюретки по 20 мл титрованного раствора алюмокалиевых квасцов в три конические колбы вместимостью 250 мл, разбавляют растворы водой до 100 мл, нейтрализуют раствором аммиака до красной окраски бумажки конго-рот. Затем добавляют по каплям раствор соляной кислоты до изменения окраски бумажки конго-рот в сиреневую и еще 8—10 капель избытка той же кислоты. К полученному раствору добавляют из бюретки 25 мл раствора трилона Б, нагревают до кипения, приливают 10 мл ацетатного буферного раствора, 6—7 капель сульфосалицилового индикатора, охлаждают до комнатной температуры и титруют раствором хлорного железа до появления золотисто-оранжевой окраски, устойчивой в течение 1 мин.
Титр раствора трилона Б, выраженный в граммах А12О3 (Т А1,о3), вычисляют по среднему арифметическому результату из трех титрований по формуле
„ 20 . б
где V — объем раствора хлорного железа, пошедший на титрование, мл;
б — титр раствора алюмокалиевых квасцов, выраженный в г А12О3;
— коэффициент, выражающий соотношение между концентрациями растворов трилона Б и хлорного железа.
2.9.2. Определение содержания окиси железа
Навеску 0,5 г цемента помещают в коническую колбу вместимостью 250 мл, содержащую 15—20 мл воды, перемешивают, приливают 5—7 мл соляной кислоты, нагревают на плитке до полного разложения цемента, избегая сильного кипения, во избежание улетучивания хлорного железа. При наличии в цементе нерастворимых в соляной кислоте добавок — 0,5 г цемента сплавляют в платиновом тигле с 2 г плавня при 1000°С в течение 3—5 мин. Сплав растворяют в 50 мл горячего раствора соляной кислоты. Затем при любом способе разложения навески добавляют 7—10 капель азотной кислоты, разбавляют водой до 100—120 мл, слабо кипятят раствор еще 1—2 мин, после чего нейтрализуют раствором аммиака до красной окраски бумажки конго, затем добавляют раствор соляной кислоты до изменения окраски бумажки на сиреневую, после чего добавляют еще 8—10 капель избытка той же кислоты и титруют горячий раствор (около 50°С) трилоном Б в присутствии 6—7 капель сульфосалицилата натрия до исчезновения фиолетовой окраски раствора.
Раствор сохраняют для определения содержания окиси алюминия.
Содержание окиси железа (БегОз) в процентах вычисляют по формулам
в)для портландцементов и пуццолановых портландцементов
Fe2O3—
F • 7Коз - 100 м
где V — объем 0,05 м раствора трилона Б, пошедший на титрование, мл;
TFeaO,— титр 0,05 м раствора трилона Б, выраженный в г Fe2Os; М — навеска цемента, г;
б) для шлакопортландцементов и клинкеров
где 1,1114 — коэффициент пересчета FeO на Fe2O3;
% FeO — определяют по п. 2.8.
Значения остальных обозначений те же, что и в предыдущей формуле.
в) для шлаков
FeO =
V -Т
Fe*O3
•0,891,
где 0,891 —коэффициент пересчета Fe2O3 на FeO.
2*9.3* Определение содержания окиси алюминия
К оттитрованному раствору по п. 2.9.2 прибавляют такое количество трилона Б, чтобы его хватило на полное связывание предполагаемого количества окиси алюминия в комплекс и остался еще избыток около 10 мл. Количество добавляемого раствора трилона Б в миллилитрах (С) вычисляют по формуле
/-» М • %А1гОа | | а
тА1зОэ-100 +10’
где М — навеска цемента, г;
%А12О3— предполагаемое содержание окиси алюминия в цементе, %;
Т ai,o3 — титр 0,05 м раствора трилона Б, выраженный в г А12Оз. После добавления трилона Б раствор нагревают до кипения, приливают 10 мл ацетатного буферного раствора, охлаждают до комнатной температуры и титруют раствором хлорного железа до появления золотисто-оранжевой окраски, не исчезающей в течение 1 мин.
Содержание окиси алюминия (А12Оз) в процентах вычисляют по формуле где С — объем добавленного раствора трилона Б, мл;
А12О3 =
(C-Ь*) »TAlsOa* 100
V — объем раствора хлорного железа, пошедший на обратное титрование, мл;
К — коэффициент, выражающий соотношение между концентрациями растворов трилона Б и хлорного железа; Taibo3 —титр 0,05 м раствора трилона Б, выраженный в г А12Оз; М — навеска цемента, г.
Примечание. Для определения окисей алюминия и железа может быть использован осадок гидроокисей, полученный по п. 2.6, после его растворения в соляной кислоте по вышеизложенной методике.
2.10. Определение содержания серного ангидрида и сульфидной серы
2.10.1. Применяемые реактивы и растворы
Плавень; готовят по п. 2.9.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее растворы 1 :3 и 1:9.
Аммиак водный по ГОСТ 3760—79, 10%-ный раствор.
Барий хлористый по ГОСТ 4108—72, 4%-ный раствор. Серебро азотнокислое по ГОСТ 1277—75, 1%-ный раствор.
2.10.2. Проведение анализа
Навеску 0,5 г цемента обрабатывают 50 мл раствора соляной кислоты 1 :9 в стакане вместимостью 300 мл. Содержимое стакана нагревают и кипятят на плитке 3—5 мин до полного разложения цемента. Горячий раствор фильтруют через неплотный фильтр. Осадок промывают 8—10 раз горячей водой и отбрасывают. При наличии в цементе сульфидной серы или нерастворимых в соляной кислоте добавок для полного ее определения в виде серного ангидрида— 0,5 г цемента сплавляют в закрытом крышкой платиновом тигле с 2 г плавня при 950—1000° С в течение 3—5 мин. Плав растворяют в 40—50 мл горячего раствора соляной кислоты 1 :3. Затем, при любом способе разложения навески, раствор или фильтрат разбавляют водой до объема не менее 200—300 мл и нейтрализуют раствором аммиака до появления легкой мути, которую растворяют несколькими каплями соляной кислоты. К прозрачному раствору прибавляют еще 2 мл соляной кислоты, нагревают раствор до кипения и осаждают серный ангидрид 25 мл кипящего раствора хлористого бария. Раствор кипятят на плитке в течение 5 мин при постоянном помешивании, оставляют в покое до просветления жидкости над осадком, затем фильтруют через двойной плотный фильтр и промывают горячей водой до исчезновения реакции на ион хлора. Осадок с фильтром переносят в предварительно прокаленный и взвешенный фарфоровый тигель, слегка подсушивают на плитке, озоляют и прокаливают в муфельной печи в течение 20—30 мин при температуре 800—850° С.
Тигель охлаждают в эксикаторе и взвешивают. Прокаливание повторяют до получения постоянной массы.
Содержание серного ангидрида (SO3) в процентах вычисляют по формуле где Mt — привес тигля, г;
SO3=
Мд *0.343» 100
М — навеска цемента, г;
0,343 — коэффициент пересчета сернокислого бария на серный ангидрид.
При анализе шлакопортландцементов, содержащих сульфатную и сульфидную серу, необходимо дважды определить количество серного ангидрида в цементе;
а) после растворения навески в соляной кислоте; выделяющийся при этом сероводород улетает и не мешает определению сульфатной серы;
б) после сплавления навески с плавнем; при этом сульфидная сера полностью окисляется в сульфатную и общее содержание серы в виде серного ангидрида определяют, как изложено выше.
Содержание в шлакопортландцементах сульфидной серы (S) в процентах вычисляют по формуле
S = (SO3 SO3 раств) •
где SO3O6ni — суммарное содержание серы в виде SO3 после окисления сульфидной серы в %;
SO3 раств — содержание сульфатной серы в %;
0,4 — коэффициент пересчета сульфатной серы на сульфидную.
Примечание. Содержание сульфидной серы в шлаках может быть определено по ГОСТ 9552—76.
2.11. Определение содержания серного ангидрида катионитовым методом
Принцип метода. Для полного растворения гипса пробу цемента сначала взбалтывают с водой, а затем нерастворимый остаток промывают борной кислотой. При наличии в гипсе труднорастворимых форм сульфата кальция пробу цемента взбалтывают с водой в смеси с катионитом в Н-форме, а затем, как и в первом случае, нерастворимый остаток промывают борной кислотой. При этом растворяется весь гипс и частично силикаты кальция.
При пропускании фильтрата через катионитовую колонку присутствующие в нем катионы поглощаются катионитовой смолой, а образовавшаяся в результате ионного обмена серная кислота от-титровывается щелочью.
Находящиеся в растворе борная и кремниевая кислоты, а также сероводородная кислота, в случае наличия сульфидов, не титруются щелочью в присутствии индикатора метилового оранжевого и не мешают определению содержания сульфатной серы, обусловленного наличием гипса.
2.11.1. Применяемые реактивы, растворы и аппаратура
Кислота борная по ГОСТ 9656—75, 5%-ный раствор.
Кислота щавелевая по ГОСТ 22180—76, 10%-ный раствор или аммоний щавелевокислый по ГОСТ 5712—78, 4%-ный раствор.
Аммиак водный по ГОСТ 3760—79, 10%-ный раствор.
Натрия гидрат окиси по ГОСТ 4328—77, 0,1 н титрованный раствор.
Метиловый оранжевый (индикатор) по ГОСТ 10816—64, 0,1‘%-ный раствор.
Кислота соляная по ГОСТ 3118—77, раствор 1 :3.
Катионитовая смола КУ-1, СБС, СБСР, КУ-2, СДВ и др. в Н-форме; готовят следующим образом: катионитовую смолу (катионит) после 6—8 ч замачивания в воде помещают в воронку, в нижней части которой находится стеклянная вата. Промывают катионит 3—4 раза раствором соляной кислоты для насыщения его водородными ионами, после чего промывают дистиллированной водой до исчезновения кислой реакции промывных вод (проба индикатором метиловым оранжевым). Хранят влажный катионит в Н-форме в банке с пробкой.
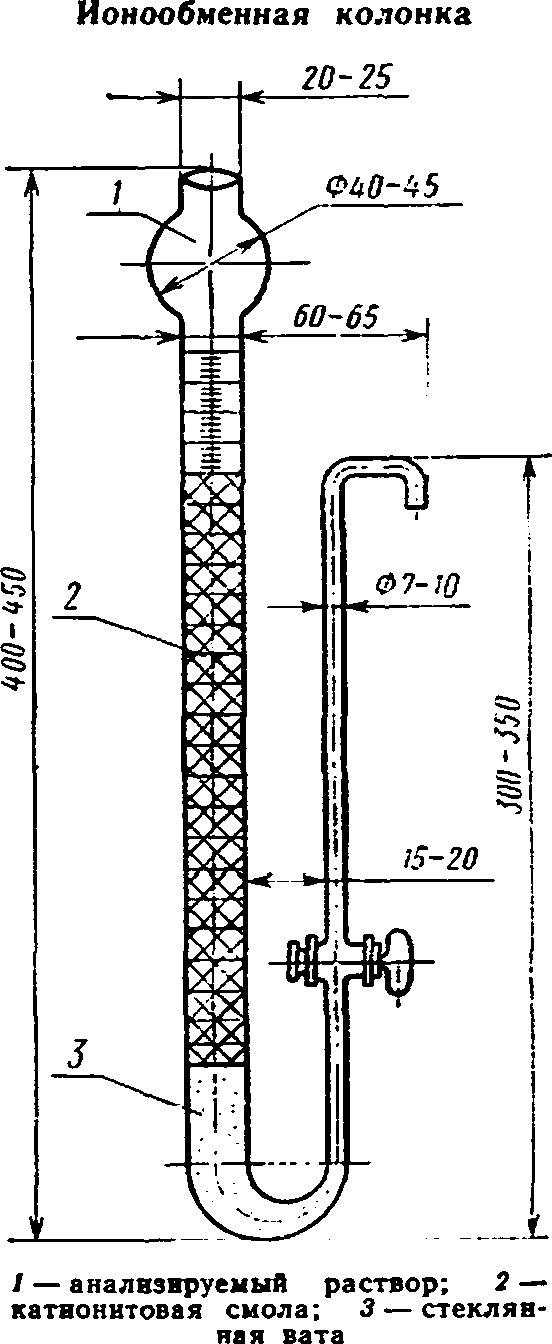
Черт. 2
Ионообменная колонка с катионитовой смолой КУ-1, СБС, СБСР, КУ-2, СДВ и др. (черт. 2).
реакции и нагревают. Если можно продолжать
Ионообменную колонку заполняют катионитовой смолой (катионитом) в Н-форме, отмытой от свободной кислоты. Смолу переносят в колонку маленькими порциями. В нижней части колонки помещают стеклянную вату, поверх нее смолу, а над ней стеклянные бусинки или вату, которые не дают смоле взмучиваться при наливании растворов. Нужно следить за тем, чтобы зерна катионита ложились плотно друг к другу. Пузырьки и прослойки воздуха уменьшают обменную емкость катионита. Колонку заполняют им на 0,5 см ниже уровня выходного отверстия боковой трубки. После 4—6 определений или по окончании рабочего дня катионит регенерируют раствором соляной кислоты и 2—3 раза промывают водой. Перед началом нового определения после регенерации катионита необходимо снова промыть колонку до отрицательной реакции на кислоту. Для выяснения возможности продолжения использования колонки без регенерации катионита проводят испытание оттитрованного фильтрата на присутствие ионов кальция. Для этого к раствору после определения содержания серного ангидрида добавляют 5—6 мл щавелевокислого аммония или щавелевой кислоты и раствора аммиака до нейтральной раствор остается прозрачным, колонкой пользоваться. Помутнение раствора указывает на необходимость немедленной регенерации колонки и повторения предыдущего анализа.
2.11.2. Проведение анализа
Навеску 0,5 г цемента помещают в сухой стакан вместимостью 150 мл и обрабатывают 25 мл воды при непрерывном перемешивании в течение 10 мин. При наличии в цементе малорастворимых форм сульфата кальция навеску 0,5 г цемента смешивают с 3 г катионита в Н-форме, обрабатывают 25 мл воды при непрерывном перемешивании в течение 10 мин. Раствору, полученному по первому или второму способу, дают отстояться до просветления и фильтруют через неплотный фильтр. Нерастворимый остаток промывают раствором борной кислоты 3—4 раза декантацией и 4—5 раз на фильтре. Через отмытую от кислоты колонку пропускают основной раствор и промывные воды небольшими порциями по мере поступления фильтрата в подставленный стаканчик со скоростью около 4 мл/мин.
После этого катионит промывают 2—3 раза водой. Фильтрат с промывными водами собирают в коническую колбу вместимостью 250 мл. Общий объем раствора после пропускания через колонку составляет около 100 мл. Полученный после катионного обмена раствор титруют раствором едкого натра по индикатору метиловому оранжевому.
Содержание серного ангидрида (SO3) в процентах вычисляют по формуле
so _ У* 0,004 . я - 100
3 _ м
где V — объем 0,1 н раствора щелочи, пошедший на титрование серной кислоты, мл;
К — поправка к титру 0,1 н раствора щелочи;
М — навеска цемента, г;
0,004 — количество серного ангидрида, соответствующее 1 мл точно 0,1 н раствора щелочи, г.
Определение содержания серного ангидрида в гипсе следует выполнять из навески 0,1 г по методике для малорастворимых форм сульфата кальция.
Примечания:
1. Результаты определения содержания серного ангидрида катионитовым методом должны периодически проверяться весовым методом по п. 2.10.
2. В случае отсутствия колонки разрешается применять обычную бюретку на 50 мл. При этом необходимо следить за тем, чтобы уровень жидкости в бюретке всегда был выше слоя катионитовой смолы (катионит).
3. Для регенерации катионита, смешанного с цементом, его отмывают от цемента водой на сите 02 мм и снова переводят на Н-форму с помощью раствора соляной кислоты.
2.12. Определение содержания окисей натрия и калия пламеннофотометрическим методом
Принцип метода. Основой пламеннофотометрического метода является непосредственное измерение интенсивности спектрального излучения элементов, определяемых из анализируемых растворов, вводимых в пламя как в источник возбуждения. Фотометрические измерения величины излучения элементов, термически возбуждаемых в пламени, позволяют количественно определять их содержание в анализируемых растворах. Излучение натрия с длиной волны 5890—5895А и калия 7665—7699А выделяется интерференционными светофильтрами с максимумами пропускания, совпадающими с длинами волн измеряемых излучений.
Присутствие в растворе алюминия, железа и магния практически не влияет на определение содержания щелочных окислов в цементах; натрий и калий также не оказывают влияния на точность определения каждого из них.
Присутствие в растворе кальция приводит к завышению результатов определения натрия вследствие недостаточной монохроматичности светофильтра, пропускающего не только излучение натрия, но и частично налагающееся на него излучение кальция в области 5500—6150А.
Для исключения мешающего влияния кальция при работе с ацетиленовым пламенем учитывают и компенсируют фототок, вызванный его излучением; при работе с пропан-бутановым пламенем переводят кальций в труднолетучее и малодиссоциируемое соединение— фосфат кальция. В последнем случае в эталонные и анализируемые растворы вводят фосфат аммония.
2.12.1. Применяемые реактивы, растворы и аппаратура
Кислота фтористоводородная (плавиковая) по ГОСТ 10484—78, 40%-ный раствор.
Кислота серная по ГОСТ 4204—77, плотностью 1,84.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 1 :3.
Аммоний фтористый по ГОСТ 4518—75.
Аммоний фосфорнокислый однозамещенный по ГОСТ 3771—74.
Смесь для сплавления; готовят следующим образом: 4 весовые части лития углекислого по МРТУ 6—09—4806—67 и 6 весовых частей борной кислоты по ГОСТ 9656—75 тщательно смешивают в фарфоровой ступке. Полученную смесь высушивают в сушильном шкафу при температуре 160—200° С в течение 2 ч, затем растирают в фарфоровой ступке и смешивают с 1 весовой частью окислителя— аммония азотнокислого по ГОСТ 22867—77. Если такая смесь при сплавлении вспучивается или разбрызгивается, то ее следует еще раз высушить и при растирании снова добавить окислитель.
Раствор для компенсации окиси кальция, содержащий около 1000 мг окиси кальция в 1 л воды; готовят следующим образом: навеску 2 г кальция хлористого плавленого по ГОСТ 4460—77 растворяют в 1 л воды и тщательно перемешивают.
Натрий хлористый безводный по ГОСТ 4233—77, 0,4 н раствор; готовят из фиксанала.
Калий хлористый безводный по ГОСТ 4234—77, 0,4 н раствор; готовят из фиксанала.
Стандартный раствор, содержащий 1000 мг Na2O и 1000 мг КгО в 1 л; готовят следующим образом: спускают из бюреток 40,35 мл 0,4 н раствора хлористого натрия и 26,55 мл 0,4 н раствора хлористого калия в мерную колбу вместимостью 500 мл, разбавляют водой до метки и хорошо перемешивают.
Пламенный фотометр.
Магнитная мешалка с подогревом.
Построение градуировочного графика. График строят, используя стандартный раствор, по серии эталонных растворов с различной концентрацией щелочных окислов и постоянной концентрацией всех реагентов, содержащихся в фотометрируемом растворе образца.
Эталонные растворы, содержащие по 1; 2; 4; 8; 10; 20; 40; 80 и 100 мг/л окисей натрия и калия, готовят следующим образом. В мерные колбы вместимостью 500 мл последовательно приливают 0,5; 1; 2; 4; 5; 10; 20; 40; 50 мл стандартного раствора. В каждую из колб добавляют по 2 мл соляной кислоты и 0,5 г соли однозамещенного фосфорнокислого аммония в случае разложения пробы смесью плавиковой и серной кислот, либо по 1 г смеси для сплавления, по 50 мл раствора соляной кислоты и по 0,5 г соли однозамещенного фосфорнокислого аммония в случае сплавления анализируемых образцов со смесью углекислого лития и борной кислоты. Растворы в колбах разбавляют водой до метки и хорошо перемешивают. Эталонные растворы наливают в фарфоровые тигли или химические стаканчики вместимостью 50 мл и производят замеры на пламенном фотометре в соответствии с имеющейся при нем инструкцией.
Первым пропускают эталон с концентрацией 100 мг/л окисей натрия и калия и устанавливают диафрагму на светофильтре таким образом, чтобы указатель гальванометра был на максимальном делении шкалы. Во время всех дальнейших измерений положение диафрагмы не меняют. Перед дальнейшими измерениями тщательно промывают распылитель водой до установки указателя гальванометра на нулевое деление шкалы. Затем последовательно замеряют интенсивности излучений эталонных проб с концентрациями щелочных окислов от 1 до 100 мг/л. Эта операция сначала выполняется для натрия с соответствующим светофильтром, а затем— для калия. По полученным показаниям гальванометра и концентрациям эталонных растворов строят градуировочные графики для окисей натрия и калия, откладывая по осн абсцисс концентрации растворов в мг/л, а по оси ординат — соответствующие им отсчеты по шкале гальванометра.
2.12.2. Определение содержания окисей натрия и калия разложением пробы в смеси серной и плавиковой кислот
Навеску 0,5 г цемента смачивают в платиновом тигле или чашке несколькими каплями воды, прибавляют 20 капель серной кислоты и 10—12 мл плавиковой кислоты (при отсутствии плавиковой кислоты можно брать 0,3 г соли фтористого аммония).
Осторожно перемешивая содержимое тигля платиновым шпателем и, не вынимая его, переносят тигель на нераскаленную песочную баню. При периодическом перемешивании платиновым шпателем отгоняют фторид кремния и избыток фтористого водорода. При появлении белых паров серного ангидрида переносят тигель на более горячее место песочной бани и нагревают до полного удаления серного ангидрида. К сухому остатку приливают 2 мл соляной кислоты и тщательно обрабатывают горячей водой в тигле, раздавливая сплющенным концом стеклянной палочки все комочки. Раствор фильтрудот через плотный фильтр в мерную колбу вместимостью 500 мл, промывая горячей водой тигель и осадок на фильтре 10—12 раз. Раствор в колбе охлаждают, доливают до метки водой и хорошо перемешивают.
Фотометрирование анализируемых образцов проводится так же, как это описано при построении графика. Проведение анализа складывается из замеров интенсивностей излучения Na2O и К2О в эталонных и анализируемых растворах и построения графиков.
Содержание Na2O или К2О (R2O) в процентах вычисляют по формуле
R О =
2 м • 1000 . 1000 ’
где All — количество К2О или Na2O, найденное по графику, мг/л; V—общий объем анализируемого раствора, мл;
М — навеска цемента, г.
2.12.3. Определение содержания окисей натрия и калия сплавлением пробы с борно-литиевой смесью
Навеску цемента 0,5 г помещают в платиновый тигель, тщательно перемешивают с 1 г смеси для сплавления и сплавляют в муфельной печи при температуре 900—950е С в течение 5—7 мин. Сплав охлаждают на воздухе и растворяют в 50 мл раствора соляной кислоты на магнитной мешалке с подогревом в течение 20—25 мин. Раствор переносят в мерную колбу вглестимостью 500 мл, охлаждают, добавляют 0,5 г соли однозамещенного фосфорнокислого аммония, разбавляют водой до метки, тщательно перемешивают, фотометрируют и вычисляют содержание Na2O и К2О в процентах по п. 2.12.2.
Примечание. При работе с ацетиленовым пламенем фосфат аммония в растворы не добавляют, а применяют оптическую компенсацию помех от кальция
2.13. Определение содержания свободной окиси кальция в клинкере
2.13.1. Применяемые реактивы, и растворы
Спирт этиловый ректификованный по ГОСТ 5962—67; спирт этиловый синтетический по ГОСТ 11547—80 или спирт гидролизный технический по ГОСТ 17299—78, готовят абсолютный спирт следующим образом: в круглодонную колбу вместимостью 3 л насыпают до половины свежую, хорошо обожженную известь и заливают более половины колбы этиловым спиртом. Спирт настаивают над известью в течение 1—2 суток. Колба должна быть плотно закрыта пробкой с хлоркальциевой трубкой. По истечении этого времени колбу соединяют с обратным холодильником, ставят на кипящую водяную баню и кипятят спирт в течение 5—6 ч. Вследствие легкой воспламеняемости спирта нагревание на водяной бане должно производиться электрическим прибором. По окончании кипячения снимают обратный холодильник и закрывают колбу каучуковой пробкой с двумя отверстиями. В одно отверстие вставляют термометр на 100° С, а в другое — изогнутую трубку, при помощи которой присоединяют колбу к холодильнику Либиха, и отгоняют спирт, производя нагревание на кипящей водяной бане.
Дистиллят собирают в сухую колбу вместимостью 1 л, закрытую пробкой с двумя отверстиями. В одно отверстие вставляют форштосс, при помощи которого колба соединяется с холодильником, во второе — хлоркальциевую трубку. Первыми 15—20 мл перегнанного спирта споласкивают приемник. Последние порции отгона (около 20 мл) также не применяют для приготовления раствора.
Полученный безводный спирт охлаждают холодной водой до 15° С и проверяют спиртометром. Если спирт окажется не 99—100-градусным, его вторично настаивают над прокаленной известью, как указано выше, и отгоняют.
Полученный безводный спирт сохраняют в склянках, закрывают их пробками и парафинируют.
Глицерин по ГОСТ 6259—75; готовят безводный глицерин следующим образом: 250—300 мл глицерина наливают в стакан вместимостью 500 мл и нагревают в течение 3 ч при температуре 160—170° С. Нагревание производят на электрической плите с асбестовой сеткой. Температуру проверяют термометром, опущенным в глицерин. При нагревании глицерин может слегка пожелтеть, но это не мешает определению. Обезвоженный и охлажденный глицерин переливают в тщательно высушенную колбу вместимостью 250—300 мл и плотно закрывают каучуковой пробкой.
Спирто-глицериновый растворитель; готовят следующим образом: 200 мл безводного глицерина наливают в коническую колбу или стакан, нагревают до температуры 100—125° С, прибавляют 15 г высушенного при температуре 130° С хлористого бария и растворяют его в глицерине. Затем раствор охлаждают и прибавляют 1 л абсолютного спирта и около 0,1 г фенолфталеина по ГОСТ 5850—72.
Обычно спирто-глицериновый растворитель бывает слегка кислым, поэтому добавляют 0,01 и спиртовой раствор едкого натра до слабощелочной реакции (бледно-розовая окраска раствора). Если раствор окажется щелочным (ярко-розовая окраска), его нейтрализуют 0,1 н спиртовым раствором бензойной кислоты до слабощелочной реакции. Спирто-глицериновый растворитель сохраняют в склянке с плотно закрытой резиновой пробкой.
Кислота бензойная по ГОСТ 10521—78, 0,1 и спиртовой раствор. Бензойную кислоту сушат в течение 24 ч в эксикаторе над серной кислотой и растворяют 12,3 г в 1 л абсолютного спирта.
Для установки титра 0,1 н спиртового раствора бензойной кислоты применяют свежепрокаленную окись кальция, полученную прокаливанием кальция углекислого по ГОСТ 4530—76 при температуре 950—1000° С в течение 2—3 ч. Тщательно растирают полученную окись кальция, переносят ее в тот же тигель, вновь прокаливают в течение 30 мин и охлаждают в эксикаторе над натронной известью.
В сухую коническую колбу вместимостью 150 мл наливают 30 мл спирто-глицеринового растворителя, затем быстро берут навеску 0,03—0,04 г смежепрокаленной окиси кальция (по разности в массе в закрытом стаканчике) и вносят ее в колбу, куда добавляют 1 г предварительно промытого (соляной кислотой и водой) и прокаленного крупнозернистого кварцевого песка, хорошо взбалтывают и присоединяют колбу к обратному холодильнику. Нагревают до кипения на электрической плитке с асбестовой сеткой и кипятят до появления интенсивно-розовой окраски раствора. Затем колбу отсоединяют от холодильника и тотчас же титруют ее содержимое 0,1 н спиртовым раствором бензойной кислоты до слаборозовой окраски. Снова присоединяют колбу к обратному холодильнику и кипятят раствор. Нагревание и титрование раствора производят до исчезновения окраски, не появляющейся при последующем нагревании в течение 20—30 мин. Титрование считается законченным, если при непрерывном кипячении в течение 30 мин не появится слаборозовая окраска раствора.
Количество окиси кальция, соответствующее 1 мл 0,1 н спиртового раствора бензойной кислоты в граммах (7сао), вычисляют по формуле
/г М
где М — навеска окиси кальция, г;
V — объем 0,1 н раствора бензойной кислоты, пошедший на титрование, мл.
Установка титра считается законченной, если результаты трех титрований дают расхождения^ не превышающие 0,00005 г окиси кальция.
2.13.2. Проведение анализа
0,1—0,5 г '(в зависимости от содержания СаОсвоб) свежеиз-мельченного клинкера помещают в коническую колбу вместимостью 150 мл и прибавляют 30 мл спирто-глицеринового растворителя. Содержимое колбы хорошо взбалтывают. Колбу присоединяют к обратному холодильнику и кипятят до появления розовой окраски раствора. Затем отсоединяют колбу от холодильника и тотчас же титруют горячий раствор 0,1 н спиртовым раствором бензойной кислоты. Нагревание и титрование производят до исчезновения окраски, не появляющейся при последующем нагревании в течение 20—30 мин. После этого титрование считают законченным.
Содержание свободной окиси кальция (СаОСВоб) в процентах вычисляют по формуле
„ У • ГСаО • 100
^аОсзоб--—-----,
где V — объем 0,1 н раствора бензойной кислоты, пошедшей на титрование, мл;
Тсао — количество окиси кальция, соответствующее 1 мл 0,1 н раствора бензойной кислоты, г;
Л1 — навеска цемента, г.
Примечания:
1. Отходы спирто-г.ишеринового растворителя собирают в банку или склянку с пробкой, затем отгоняют при температуре 78° С, охлаждают безводный спирт холодной водой до 15° С, проверяют спиртомером. При этом спирт, как правило, получается абсолютный и может быть использован для работы.
2. Следует иметь в виду, что при анализе лежалого клинкера, наряду с СаОсвоб по изложенному методу определяется и содержание гидрата окиси кальция.
3. При определении содержания свободной извести в гидратированных цементах или клинкерах полученный результат СаОсвоб необходимо пересчитать на гидрат окиси кальция умножением на коэффициент .1,32.
2.14. Определение содержания окиси бария в барийсодержащих портландцементах
Для определения содержания основных компонентов навеску барийсодержащего цемента сплавляют со смесью соды и буры и выщелачивают теплым раствором соляной кислоты. Возможное частичное выпадение осадка сернокислого бария не влияет на выполнение анализа. Присутствующая в цементе кремнекислота в
данных условиях находится в растворимой форме и не мешает весовому определению содержания окиси бария.
2.14.1. Применяемые реактивы и растворы
Плавень; готовят по п. 2.9.
Кислота серная по ГОСТ 4204—77, 5%-ный раствор.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 1 :3.
Аммиак водный по ГОСТ 3760—79, 10%-ный раствор.
Серебро азотнокислое по ГОСТ 1277—75, 1%-ный раствор.
2.14.2. Проведение анализа
Навеску 0,5 г цемента сплавляют с 2 г плавня, после охлаждения сплав выщелачивают 40—50 мл теплого раствора соляной кислоты в стакане вместимостью 300—400 мл до полного растворения. Полученный раствор разбавляют водой до 200—250 мл, не обращая внимания на выпавший осадок сернокислого бария, нейтрализуют аммиаком до появления легкой мути гидроокисей, которую растворяют несколькими каплями соляной кислоты и добавляют избыток этой же кислоты в 2 мл.
Затем раствор нагревают до кипения, добавляют 15 мл горячего раствора серной кислоты и кипятят в течение 2—3 мин. Раствор с осадком выдерживают в теплом месте до тех пор, пока жидкость над осадком не станет прозрачной, затем фильтруют через двойной плотный фильтр и промывают горячей водой до исчезновения реакции на ион хлора. Осадок вместе с фильтром переносят в предварительно прокаленный и взвешенный фарфоровый тигель, подсушивают, озоляют и прокаливают в течение 20—30 мин в муфельной печи при температуре 800—850° С. Тигель охлаждают в эксикаторе и взвешивают. Прокаливание повторяют до получения постоянной массы.
Содержание окиси бария (ВаО) в процентах вычисляют по формуле
ВаО = ' 100 ,
м
где Mi — масса осадка BaSO4, г;
М — сухая навеска цемента, г;
0,657 — коэффициент пересчета BaSO4 на ВаО.
Фильтрат после отделения сернокислого бария переводят в мерную колбу вместимостью 250 мл, разбавляют водой до метки, хорошо перемешивают и используют для комплексонометрического определения окиси кальция по п. 2.7.
2.15. Определение содержания серного ангидрида в барийсодержащих портландцементах
2.15.1. Применяемые реактивы и растворы
Барий хлористый по ГОСТ 4108—72, 4%-ный раствор.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и раствор ее 1 :3.
Остальные реактивы — по п. 2.10.
2.15.2. Проведение анализа
Навеску 0,5 г цемента сплавляют с 2 г плавня, после охлаждения сплава его выщелачивают 40—50 мл теплого раствора соляной кислоты в стакане вместимостью 300—400 мл. После полного растворения сплава раствор разбавляют водой до 250—300 мл, не обращая внимания на выпавший осадок сернокислого бария, раствор нейтрализуют аммиаком до появления легкой мути гидроокисей, которую растворяют несколькими каплями соляной кислоты и добавляют избыток этой же кислоты в 2 мл. Затем раствор нагревают до кипения и добавляют 25 мл кипящего раствора хлористого бария для полноты осаждения серного ангидрида. Все остальные операции и вычисление содержания серного ангидрида производят по п. 2.10.
Примечание. Содержание S1O2» А12О3, Fe2O3 и MgO определяют колориметрическим методом по пп. 3.1, 32, 3.3, 3.7 из аликвотных частей раствора, полученного сплавлением 0,1 г цемента с 1 г плавня, последующего выщелачивания сплава 100 мл теплого раствора соляной кислоты 1 :3. Раствор переводят в мерную колбу вместимостью 500 мл, доливают до метки водой, хорошо перемешивают и выдерживают 20—30 мин до полного просветления раствора над осадком; аликвотные части раствора отбирают осторожно, не взмучивая осадка.
3. ФОТОКОЛОРИМЕТРИЧЕСКИЕ МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА
3.1. Определение содержания двуокиси кремния
Принцип метода. Колориметрическое определение SiO2 основано на образовании желтого комплекса кремнемолибденовой гетерополикислоты, в результате реакции кремневой кислоты с молибдатом аммония в кислой среде.
Для повышения чувствительности определения желтый кремнемолибденовый комплекс переводят в синий путем восстановления аскорбиновой кислоты, либо метолово-сульфитным раствором.
3.1.1. Применяемые реактивы, растворы и аппаратура
Плавень; готовят по п. 2.9.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и раствор ее 1 :3.
Раствор восстановителя 1; готовят следующим образом: 1 г аскорбиновой кислоты и 5 г лимонной кислоты по ГОСТ 3652—69 растворяют в 50 мл воды, отфильтровывают и разбавляют водой до 100 мл.
Раствор восстановителя 2; готовят следующим образом: 20 г метола (пара-метиламиносульфата) по ГОСТ 25664—83, 12,5 г лимонной кислоты по ГОСТ 3652—69 и 12 г сернистокислого натрия (натрий сульфита) безводного по ГОСТ 195—77 растворяют в 800 мл воды и доливают водой до 1 л.
Аммоний молибденовокислый по ГОСТ 3765—78, 5%-ный водный раствор.
Стандартный раствор готовят на основе образца сырьевой смеси, химический состав которой определен в разных лабораториях по настоящему стандарту. Этот образец в дальнейшем именуется стандартным. 0,15 г стандартного образца смешивают с 1 г плавня и сплавляют в платиновом тигле, закрытом крышкой, при температуре 1000° С в течение 3—5 мин. Охлажденный тигель помещают в стакан, содержащий 100 мл раствора соляной кислоты, и выщелачивают сплав при постоянном перемешивании до полного растворения. Раствор количественно переносят в мерную колбу вместимостью 500 мл, доливают до метки водой и хорошо перемешивают. Такой раствор можно использовать в течение 1—2 месяцев для построения или проверки градуировочных графиков по определению содержания двуокиси кремния, окисей алюминия, железа, магния и фототрилонометрического определения кальция.
Фотоэлектроколориметр.
Построение градуировочного графика. Градуировочный график строят по серии эталонных растворов с различной концентрацией кремнекислоты, приготовленных из стандартного раствора. В три мерные колбы вместимостью 100 мл последовательно приливают 3; 5; 7 мл стандартного раствора, добавляют в каждую примерно по 25 мл воды, 5 мл раствора молибдата аммония, перемешивают и дают постоять 10 мин для полного образования желтого кремнемолибденового комплекса. Затем добавляют по 5 мл раствора восстановителя 1, либо по 20 мл восстановителя 2, разбавляют водой до метки, хорошо перемешивают, дают постоять 15 мин для получения синего комплекса и колориметрируют в кюветах размером 10 мм с красным светофильтром (Х=656—680 нм). Одну кювету наполняют Эталонным раствором, две другие такого же размера — дистиллированной водой и определяют оптические плотности растворов в соответствии с инструкцией, приложенной к фотоколориметру. По полученным результатам определения оптической плотности и известной концентрации эталонных растворов строят градуировочный график для данного размера кювет, откладывая по оси абсцисс концентрацию кремнекислоты в миллиграммах в 100 мл раствора, а по оси ординат — соответствующую им оптическую плотность.
Градуировочный график можно строить, откладывая по оси абсцисс содержание SiO2 в процентах, а по оси ординат — соответствующую оптическую плотность раствора. При этом аликвотную часть раствора в количестве 5 мл принимают за основную и концентрацию SiO2 в ней приравнивают процентному содержанию SiO2 в стандартном образце. В двух других аликвотных частях концентрацию SiO2 соответственно рассчитывают.
На основе градуировочных графиков могут быть составлены расчетные таблицы, где каждому значению оптической плотности будет соответствовать определенная концентрация искомого окисла.
3.1.2. Проведение анализа
Навеску 0,1 г цемента сплавляют с 1 г плавня. Сплав растворяют, непрерывно перемешивая его в 100 мл раствора соляной кислоты, и переносят количественно прозрачный раствор в мерную колбу вместимостью 500 мл, как указано в описании построения градуировочного графика. Полученный раствор служит для колориметрического определения содержания двуокиси кремния, окисей железа, алюминия, магния и фототрилонометрического определения кальция.
Для определения содержания двуокиси кремния последовательно отбирают в две мерные колбы вместимостью 100 мл по 5 мл анализируемого и 5 мл стандартного растворов, добавляют около 25 мл воды, 5 мл раствора молибдата аммония и производят все дальнейшие операции, вплоть до колориметрирования, как указано в описании построения градуировочного графика. Затем вводят поправку на изменение условий колориметрирования по сравнению с условиями построения графика по измеренной оптической плотности эталонного раствора. Поправка вносится с обратным знаком, т. е. если оптическая плотность эталонного раствора увеличилась на 0,007, то эта величина отнимается от оптической плотности раствора цемента и наоборот. Затем определяют по графику содержание двуокиси кремния в миллиграммах с учетом введенной поправки.
Содержание двуокиси кремния (SiO2) в процентах вычисляют по формуле
SiO2 = M1'V' 1С? ,
2 м • vt • 1000’
где All — количество двуокиси кремния, найденное по графику, мг;
V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл;
М — навеска цемента, г.
При выражении концентраций градуировочных графиков в процентах определяемых компонентов полученные результаты для цементов увеличивают в 1,5 раза. Приведенный расчет может быть применен для всех фотоколориметрических определений, за исключением дифференциального.
При необходимости изменения навески анализируемого образца, разведения или аликвотной части, содержание двуокиси кремния (S1O2) в процентах вычисляют по формуле где М — содержание двуокиси кремния, найденное по графику,
SiOa =
м
• Ki- Л'з ’
Kt — отношение навески анализируемого образца к навеске 0,15 г;
К2 — отношение разведения 500 мл к разведению анализируемого образца;
Кз — отношение аликвотной части анализируемого раствора к стандартной аликвотной части.
3.1.3. Определение содержания двуокиси коемния дифференциальным методом
Принцип метода. Для увеличения точности определения повышенных количеств двуокиси кремния в пуццолановых портландцементах и шлакопортландцементах используют дифференциальный фотоколориметрический метод, основанный на измерении оптической плотности анализируемого раствора по отношению к оптической плотности эталонного раствора стандартного образца.
Обязательным условием этого метода является использование оптически равноценных кювет, что проверяется получением одинаковой оптической плотности при измерении одного и того же окрашенного раствора в обеих кюветах.
3.1.4. Применяемые реактивы и растворы
Стандартный раствор; готовят на основе образца сырьевой смеси из навески 0,2 г аналогично п. 3.1.1.
Остальные реактивы и растворы — по п. 3.1.1.
Построение градуировочного графика. В три мерные колбы емкостью 100 мл последовательно приливают 3; 5 и 7 мл стандартного раствора, добавляют примерно по 25 мл воды и все остальные реактивы вплоть до получения синего комплекса кремнемолибденовой гетерополикислоты, аналогичного п. 3.1. Колориметрирование производят в кюветах размером 10 мм, пользуясь красным светофильтром (Х=656—680 нм), следующим образом. Аликвотную часть раствора в количестве 5 мл считают основной и относительно нее колориметрируют остальные аликвотные части. Для этого ставят кювету с аликвотной частью в 5 мл в правый держатель на пути пучка света, а шкалу оптических плотностей устанавливают на 0,250 и путем вращения рукоятки фотометрического клина стрелку гальванометра переводят на нуль. Затем относительно основного раствора проверяют оптические плотности растворов двух других аликвотных частей (3 и 7 мл). По полученным величинам оптических плотностей и соответствующих им концентраций растворов, выраженных в миллиграммах или процентах, строят градуировочный график.
3.1.5. Проведение анализа
Навеску 0,1 г цемента сплавляют с 1 г плавня. Плав растворяют, непрерывно перемешивая его в 100 мл раствора соляной кислоты, переносят количественно прозрачный раствор в мерную колбу вместимостью 500 мл, доливают до метки водой и хорошо перемешивают.
Для определения содержания двуокиси кремния последовательно отбирают в две мерные колбы вместимостью 100 мл 5 мл анализируемого и 5 мл стандартного растворов и все остальные реактивы по п. 3.1.1, вплоть до получения синего комплекса кремнемолибденовой гетерополикислоты. Затем в две кюветы размером 10 мм наливают окрашенные эталонный и анализируемый растворы; оптическую плотность эталонного раствора устанавливают на значение шкалы 0,250 при нулевом положении стрелки гальванометра, а затем измеряют оптическую плотность анализируемого раствора.
Содержание двуокиси кремния в процентах вычисляют по ранее приведенной формуле.
При выражении концентраций градуировочных графиков в процентах определяемых компонентов полученные результаты для цементов увеличивают в два раза.
3.2. Определение содержания окиси алюминия
Принцип метода. Колориметрическое определение окиси алюминия основано на образовании окрашенного в красный цвет комплексного соединения иона алюминия с алюминоном. Наиболее устойчивая окраска комплексного соединения получается при pH раствора 4,5—4,8.
3.2.1. Применяемые реактивы, растворы и аппаратура
Алюминон по ГОСТ 9859—74, 0,1%-ный раствор; готовят следующим образом: 0,5 г алюминона растворяют в небольшом количестве воды, переносят в мерную колбу вместимостью 500 мл, разбавляют водой до метки, хорошо перемешивают, помещают в склянку из темного стекла и выдерживают в течение 4—5 суток в темном месте.
Буферный раствор с pH около 5,2—5,3; готовят следующим образом: 100 г плавленого натрия уксуснокислого по ГОСТ 199—78 или 166 г трехводной соли растворяют в 300 мл воды, раствор фильтруют, разбавляют водой примерно до 950 мл, добавляют 15 мл ледяной уксусной кислоты или 20 мл 80%-ной уксусной кислоты, хорошо перемешивают и проверяют величину pH раствора на потенциометре, pH-метре или с помощью универсальной индикаторной бумажки; при pH раствора около 5,2—5,3 его можно применять.
Кислота аскорбиновая, 0,2%-ный водный раствор.
Кислота уксусная по ГОСТ 61—75, ледяная или 80%-ная. Потенциометр или рН-метр.
Фотоэлектроколориметр.
Построение градуировочного графика. В три мерные колбы вместимостью 50 мл последовательно приливают 3; 5 и 7 мл стандартного раствора, приготовленного, как указано в п. 3.1. В связи с тем, что при определении содержания окиси алюминия особенно важно соблюдать строго определенную и одинаковую кислотность раствора, необходимо для получения оптической плотности при построении градуировочного графика и в случае колориметрирования аликвотных частей стандартного раствора объемом 3 и 7 мл сначала приравнять их кислотность к кислотности раствора, соответствующей 5 мл, поскольку это количество колориметрируется при основном определении. Для этого используют титрованные растворы 1 н НС1 и 0,25 н NaOH, имеющиеся во всех лабораториях. Отбирают дополнительно в стакан или коническую колбу 2 мл стандартного раствора, приготовленного по п. 3.1, и нейтрализуют его 0,25 н раствором NaOH по фенолфталеину. Пошедшее на нейтрализацию количество щелочи добавляют к отобранной ранее аликвотной части стандартного раствора объемом 7 мл. К аликвотной же части стандартного раствора объемом 3 мл добавляют в четыре раза меньшее количество 1 н НС1. После этого во все колбы добавляют по 1 мл раствора аскорбиновой кислоты и по 5 мл раствора алюминона, затем добавляют по 10 мл ацетатного буферного раствора, разбавляют до метки водой, хорошо перемешивают и после 15 мин выдерживания раствора колориметрируют в кюветах размером 10 мм, пользуясь зеленым светофильтром (1=530—536 нм). По полученным результатам определения оптической плотности и известной концентрации эталонных растворов строят градуировочный график, как указано в п. 3.1.
3.2.2. Проведение анализа
В две мерные колбы вместимостью 50 мл отбирают по 5 мл анализируемого и стандартного растворов, полученных по п. 3.1, вводят по 1 мл раствора аскорбиновой кислоты и по 5 мл раствора алюминона, добавляют по 10 мл ацетатного буферного раствора, разбавляют до метки водой, хорошо перемешивают и после 15 мин выдерживания колориметрируют в кюветах размером 10 мм, пользуясь зеленым светофильтром.
Содержание окиси алюминия (AI2O3) в процентах вычисляют по формуле
100
где Mi — количество A12O3, найденное по графику, мг;
V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл; М — навеска цемента, г.
3.3. Определение содержания окиси железа
Принцип метода. Колориметрическое определение содержания железа основано на образовании окрашенного в красный цвет комплексного соединения иона железа с ионом роданида или в желтый цвет с сульфосалицилатом. Роданид железа очень чувствителен к изменению температуры, он легко разлагается при освещении солнечным или искусственным светом, поэтому колориметрирование роданидного комплекса необходимо производить быстро, точно соблюдая условия анализа.
3.3.1. Применяемые реактивы, растворы и аппаратура
Аммиак водный по ГОСТ 3760—79, 25%-ный раствор.
Кислота сульфосалициловая по ГОСТ 4478—78, 25%-ный раствор.
Аммоний роданистый по СТ СЭВ 222—75, 25%-ный раствор или калий роданистый по ГОСТ 4139—75, 30%-ный раствор.
Кислота азотная по ГОСТ 4461—77, 4 н раствор; готовят следующим образом: 276 мл азотной кислоты плотностью 1,4 разбавляют водой до 1 л.
Кислота соляная по ГОСТ 3118—77, раствор 1 :3.
Фотоэлектроколориметр.
А. Роданидный метод
3.3.2. Построение градуировочного графика. В три мерные колбы вместимостью 10 мл последовательно вливают 10; 25 и 50 мл стандартного раствора, приготовленного по п. 3.1. Затем во все колбы добавляют по 10 мл раствора азотной кислоты и поочередно по 10 мл раствора роданида, разбавляют до метки водой, хорошо перемешивают и сразу же колориметрируют с синим светофильтром (Л=420—453 нм) в кюветах размером 10 мм.
Ввиду того, что роданид железа под действием света легко разлагается, при построении графика и определении железа необходимо приливать роданид калия или аммония перед измерением оптической плотности. По полученным значениям оптической плотности и известной концентрации эталонных растворов строят градуировочный график.
3.3.3. Проведение анализа
Отбирают пипеткой 25 мл анализируемого раствора, полученного по п. 3.1, переносят его в мерную колбу вместимостью 100 мл, добавляют 10 мл раствора азотной кислоты и 10 мл раствора роданида, разбавляют водой до метки, перемешивают и колориметрируют так же, как при построении графика.
Содержание окиси железа (Ре20з) в процентах вычисляют по формуле
Fe2O3=
«У - 100 м . Ух • 1000’
где Mi — количество окиси железа, найденное по графику, мг; V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл; М — навеска цемента, г.
Б. Сульфосалицилатный метод
Построение градуировочного графика. В три мерные колбы вместимостью 100 мл последовательно приливают 10; 25 и 50 мл стандартного раствора, приготовленного по п. 3.1. Затем добавляют по 15 мл раствора сульфосалициловой кислоты и по каплям раствор аммиака до получения устойчивого желтого окрашивания раствора и еще 5 мл избытка раствора аммиака. После этого растворы разбавляют до метки водой, колориметрируют в кюветах размером 10 мм с синим светофильтром (Х=420—453 нм) и по полученным результатам строят градуировочный график.
3.3.4. Проведение анализа
Определение содержания окиси железа производят из аликвотной части основного раствора 25 мл и далее поступают так же, как при построении графика.
Расчет содержания окиси железа производят по формуле, приведенной для роданидного метода.
Примечание. При определении содержания окиси железа в белом портландцементе 0,1—0,5 г цемента сплавляют с 2 г плавня, выщелачивают в 30—40 мл раствора соляной кислоты, переводят в мерную колбу вместимостью 100 мл и колориметрируют, как изложено выше,
Содержание окиси железа (Fe2O3) в процентах вычисляют по формуле, приведенной для роданидного метода.
3.4. Определение содержания двуокиси титана Принцип метода. Колориметрический метод определения титана основан на образовании в кислой среде окрашенного в желтый цвет комплексного соединения титана с перекисью водорода или в золотисто-желтый с диантипирилметаном (ДАМ).
3.4.1. Применяемые реактивы, растворы и аппаратура
Кислота соляная по ГОСТ 3118—77, раствор 1 :3.
Кислота серная по ГОСТ 4204—77, плотностью 1,84 и ее раствор 5:95,
Перекись водорода по ГОСТ 10929—76, 3%-иый раствор. Плавень; готовят по п. 2.9.
Аммиак водный по ГОСТ 3760—79, 10%-ный раствор.
Стандартный раствор соли четырехвалентного титана; готовят следующим образом: навеску 0,05 г химически чистой двуокиси титана сплавляют в платиновом тигле с 1 г плавня при температуре 950—1000° С в течение 3—5 мин и по охлаждении выщелачивают в растворе серной кислоты.
Прозрачный раствор переводят в мерную колбу вместимостью 1 л, разбавляют до метки раствором серной кислоты 5:95 и хорошо перемешивают. 1 мл стандартного раствора содержит ■ около 0,05 мг ТЮг.
Содержание двуокиси титана в 1 мл раствора в миллиграммах устанавливают весовым методом в параллельных пробах путем двукратного осаждения раствором аммиака из 100 мл приготовленного раствора, последующего прокаливания и взвешивания осадка.
Количество двуокиси титана в 1 мл раствора в миллиграммах — титр (TiO2) вычисляют по формуле
М-1000 Т1О2 =-------.
где G — масса прокаленного осадка ТЮг, г.
Диантипирилметан по МРТУ 6—09—2120—65, 5%-ный раствор в 1 н растворе соляной кислоты; готовят следующим образом: 50 г ДАМ растворяют в 300—400 мл 1 н раствора соляной кислоты, хорошо перемешивают и доводят до литра этой же кислотой.
Медь сернокислая безводная по ТУ 6—09—4525—77, 5%-ный раствор.
Кислота аскорбиновая, 0,2%-ный раствор.
Фотоэлектроколориметр.
А. Перекисный метод
Построение градуировочного графика. График строят по данным, полученным в результате колориметрирования серии эталонных растворов, приготовленных из стандартного раствора соли титана.
В четыре мерные колбы вместимостью 100 мл последовательно вливают 10; 20; 30 и 40 мл стандартного раствора соли титана с содержанием около 0,05 мг ТЮ2 в 1 мл, добавляют примерно по 50 мл раствора серной кислоты и по 3 мл перекиси водорода, разбавляют до метки раствором серной кислоты, хорошо перемешивают колориметрируют в кюветах размером 50 мм с синим светофильтром (%=420—453 нм).
Колориметрирование и построение градуировочного графика производят аналогично методам, приведенным в п. 3.1.
3.4.2. Проведение анализа
Навеску 0,5 г цемента сплавляют с 2 г плавня при температуре 950—1000° С в течение 3—5 мин. После быстрого охлаждения сплав выщелачивают 30—40 мл теплого раствора серной кислоты. Раствор переводят в мерную колбу вместимостью 250 мл, охлаждают, доливают до метки раствором серной кислоты и хорошо перемешивают.
Отбирают пипеткой 25—50 мл этого раствора, переносят в мерную колбу вместимостью 100 мл, добавляют 3 мл перекиси водорода, доливают до метки раствором серной кислоты, хорошо перемешивают и колориметрируют, как это изложено при построении градуировочного графика.
Содержание двуокиси титана (TiO2) в процентах вычисляют по формуле
’ м . • 1000*
где — количество двуокиси титана, найденное по градуировочному графику, мг;
V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл;
М — навеска цемента, г.
Б. Диантипирилметановый метод
Построение градуировочного графика. В три мерные колбы вместимостью 100 мл последовательно приливают 4; 6 и 8 мл стандартного раствора соли титана, содержащего около 0,05 мг TiO2 в 1 мл, добавляют по 10 мл раствора соляной кислоты, по две капли раствора сернокислой меди, по 10 мл аскорбиновой кислоты и по 5 мл ДАМ. Растворы доливают до метки водой, хорошо перемешивают, дают постоять 45 мин для получения золотисто-желтого комплексного соединения, после чего колориметрируют в кювете 20 мм с синим светофильтром (Х=420—453 нм).
По полученным результатам определения оптической плотности и известной концентрации строят градуировочный график для данного размера кювет.
3.4.3. Проведение анализа
Отбирают пипеткой 50 мл анализируемого раствора, полученного по п. 3.1, переносят его в мерную колбу вместимостью 100 мл, добавляют 2 капли раствора сернокислой меди, 10 мл аскорбиновой кислоты, 5 мл диантипирилметана, разбавляют водой до метки, перемешивают и через 45 мин колориметрируют в кюветах размером 20 мм, пользуясь синим светофильтром.
Расчет содержания двуокиси титана производят по формуле, приведенной для перекисного метода.
3.5. Определение содержания закиси марта н-ц а
А. Перманганатный метод
Принцип метода. Колориметрический метод определения закиси марганца основан на окислении его в азотнокислой среде надсернокислым аммонием до перманганата в присутствии азотнокислого серебра в качестве катализатора.
3.5.1. Применяемые реактивы, растворы и аппаратура
Кислота азотная по ГОСТ 4461—77, плотностью 1,4 и ее растворы 1 :4 и 1 н.
Серебро азотнокислое по ГОСТ 1277—75, 0,2%-ный раствор. Аммоний надсернокислый (персульфат) по ГОСТ 20478—75. Стандартный раствор 1; готовят на основе стандартного образца шлака 267а. Навеску образца сплавляют в платиновом тигле с 2 г плавня при температуре 950—1000° С в течение 3—5 мин, затем тигель охлаждают и помещают в стакан вместимостью 150 мл, содержащий 50 мл горячего раствора азотной кислоты 1 :4 и выщелачивают плав при постоянном перемешивании до полного растворения. В случае выпадения бурого осадка или мути четырехвалентного марганца необходимо добавлять кристаллики аскорбиновой кислоты или сернокислого гидроксиламина до обесцвечивания раствора. Полученный раствор переносят в мерную колбу вместимостью 250 мл, охлаждают, доводят до метки водой и хорошо перемешивают. 1 мл стандартного раствора содержит 0,0282 мг МпО.
Стандартный раствор 2; готовят 0,025 н титрованный раствор калия марганцовокислого по ГОСТ 20490—75 из фиксанала. 1 мл точно 0,025 н раствора КМпО4 соответствует 0,3547 мг МпО.
Плавень; готовят, как указано в п. 2.9.
Кислота аскорбиновая.
Гидроксиламин сернокислый по ГОСТ 7298—79. Фотоэлектроколориметр.
Построение градуировочного графика. График строят по данным, полученным в результате колориметрирования серии эталонных растворов, приготовленных из стандартного раствора 1 или 2. В четыре мерные колбы вместимостью 100 мл последовательно приливают 20; 30; 40 и 50 мл стандартного раствора 1, разбавляют примерно до 60 мл 1 н раствором азотной кислоты, прибавляют по 5 мл азотнокислого серебра, 2—3 г надсернокислого аммония и дают постоять на кипящей водяной бане до момента установления постоянной интенсивности окраски раствора. Если окраска раствора имеет буроватый оттенок, добавляют еще немного надсернокислого аммония и несколько миллилитров азотнокислого серебра. Раствор охлаждают до комнатной температуры» доводят его объем до метки 1 н раствором азотной кислоты и хорошо перемешивают. Необходимо, чтобы раствор в колбе имел примерно 1 н концентрацию азотной кислоты, т. е. содержал около 6,6 мл азотной кислоты в 100 мл раствора. Затем все три раствора колориметрируют в кюветах размером 10 мм, используя зеленый светофильтр (Х=530—536 нм). Колориметрирование и построение градуировочного графика производят, как указано в п. 3.1. Для построения градуировочного графика по данным, полученным по результатам колориметрирования стандартного раствора 2, в четыре мерные колбы вместимостью 100 мл последовательно приливают 1; 2; 3 и 4 мл 0,025 н раствора марганцовокислого калия, разбавляют 1 н раствором азотной кислоты до метки, перемешивают и колориметрируют.
3.5.2. Проведение анализа
Навеску 0,5 г цемента сплавляют с 2 г плавня при температуре 950—1000° С в течение 3—5 мин. После быстрого охлаждения плав выщелачивают 30 мл теплого раствора азотной кислоты 1 :4 в стакане вместимостью 150 мл, ставят на кипящую водяную баню, добавляют 5 мл раствора азотнокислого серебра, 2—3 г надсернокислого аммония и дают постоять на водяной бане до момента установления постоянной интенсивности окраски раствора и производят все дальнейшие операции вплоть до колориметрирования так, как это указано при описании построения градуировочного графика по п. 3.1. По полученной оптической плотности находят по графику концентрацию испытуемого раствора.
Содержание закиси'марганца (МпО) в процентах вычисляют по формуле
Ml. uo
Мп О =
М • 1000 ’
где Mi — количество закиси марганца, найденное по градуировочному графику, мг;
М — навеска цемента, г.
Б. Формальдоксимный метод
Принцип метода. Колориметрический метод основан на реакции двухвалентного марганца с формальдоксимом в щелочной среде при рН=9,4—9,6 с образованием окрашенного в красно-бурый цвет комплекса — формальдоксимата марганца, состав которого соответствует формуле Мп (H2C = NO)3. Мешающее влияние трехвалентного железа устраняют путем его восстановления дс двухвалентного. Для маскирования присутствующих R2O3 применяют триэтаноламин в смеси с фторидом натрия.
По данному методу можно определить содержание марганца в солянокислом растворе, приготовленном для фотометрического анализа.
3.5.3. Применяемые реактивы, растворы и аппаратура
Кислота соляная по ГОСТ 3118—77, раствор 1 :3.
Аммиак водный по ГОСТ 3760—79, 10%-ный раствор.
Кислота аскорбиновая.
Натрий сернистокислый безводный по ГОСТ 195—77, 1—2 н раствор.
Формальдоксим по МРТУ 6—09—1568—64, 2’%-ный раствор; готовят из гидроксиламина и формалина следующим образом: 4 г гидроксиламина гидрохлорида по ГОСТ 5456—79 и 4—5 мл 40% -ного формалина технического по ГОСТ 1625—75 растворяют в 250 мл воды.
Маскирующий реагент; готовят следующим образом: 10 мл триэтаноламина по МРТУ 6—09—403—67 и 5 г фтористого натрия по ГОСТ 4463—76 растворяют в 300—400 мл воды, фильтруют и разбавляют водой до 1 л.
Стандартный раствор, содержащий 0,02 мг/мл МпО; готовят из 0,1 н раствора марганцовокислого калия по ГОСТ 20490—75 с известным титром по МпО либо из 0,1 н раствора КМпО4, приготовленного из фиксанала. Количество миллилитров 0,1 н раствора марганцовокислого калия рассчитывают по формуле
0.02-V мво • 1°°° ’
где 0,02 — содержание МпО, мг/мл;
V — разведение, мл;
Гмпо — титр раствора марганцовокислого калия, выраженный в г МпО.
Рассчитанное количество 0,1 н раствора марганцовокислого калия в миллилитрах отбирают в стакан (1:3) вместимостью 200—300 мл, прибавляют 100 мл раствора соляной кислоты (1 :3) и 3—4 капли раствора сульфита натрия до обесцвечивания раствора КМпО4. Раствор кипятят до удаления запаха сернистого ангидрида, переносят в мерную колбу вместимостью 250 мл, охлаждают до комнатной температуры, доводят водой до метки и хорошо перемешивают.
Построение градуировочного графика. В четыре мерные колбы вместимостью 100 мл последовательно приливают 2; 4; 6 и 8 мл стандартного раствора, добавляют около 50 мл-воды, 5 мл раствора аммиака, 5 мл формальдоксима, доводят до метки водой, хорошо перемешивают и дают постоять 15 мин. Затем все четыре раствора колориметрируют в кюветах размером 20 мл с синим светофильтром (Х=420—453 нм) и строят градуировочный график так, как это изложено в п. З.5.А.
Фотоэлектроколориметр.
3.5.4. Проведение анализа
Отбирают пипеткой 25 мл анализируемого раствора, полученного, как указано в п. 3.1, переносят его в мерную колбу вместимостью 100 мл, разбавляют водой примерно до 50 мл, прибавляют 5 мл маскирующего реагента, 0,05 г аскорбиновой кислоты, 8 мл раствора аммиака и 5 мл раствора формальдоксима. Раствор доводят до метки водой, хорошо перемешивают и через 15 мин колориметрируют так же, как это описано при построении графика в п. 3.1.
Содержание закиси марганца (МпО) в процентах вычисляют по формуле
МпО =
- V - 100 М . • 1000*
где Afi — количество закиси марганца, найденное по градуировочному графику, мг;
V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл; М — навеска цемента, г.
3.6. Определение содержания окиси кальция фототрилонометрическим титрованием на приборах ФЭТ-УНИИЗ и ТФЛ-46—2.
Принцип метода. Принцип фототрилонометрического титрования основан на том, что в процессе титрования вблизи эквивалентной точки титруемый раствор начинает изменять свою окраску, при этом меняются величина светопоглощения раствора и величина фототока, силу которого регистрирует микроамперметр.
При титровании кальция на ФЭТ-УНИИЗ в точке эквивалентности достигается максимальная величина светопоглощения и стрелка микроамперметра останавливается.
Точка эквивалентности на приборе ТФЛ-46—2 фиксируется электронным блоком фотоабсорбциометра, сигнал с которого воздействует на электромагнитный клапан титрования, последний перекрывает магистраль, и подача титранта прекращается. На приборе ТФЛ-46—2 комплексонометрическое титрование кальция производится в две стадии:
1) автоматическое прекращение подачи титранта, фиксируемое вблизи точки эквивалентности;
2) окончательное дотитровывание раствора визуальным способом.
3.6.1. Применяемые реактивы, растворы и аппаратура
Натрия гидрат окиси по ГОСТ 4328—77 или калия гидрат окиси, 20%-ные растворы.
Маскирующий реагент; готовят по п. 3.5.3.
Кальций углекислый по ГОСТ 4530—76.
Кислотный хром темно-синий (индикатор) по ГОСТ 14091—78, 0,5%-ный водный раствор; готовят следующим образом: 0,5 г кислотного хрома темно-синего растворяют в 100 мл воды и фильтруют.
Трилон Б по ГОСТ 10652—73, 0,01 м раствор; готовят следующим образом: около 3,6 г трилона Б растворяют в 1 л воды и хорошо перемешивают. Если раствор получился мутным, его фильтруют. Титр раствора устанавливают по углекислому кальцию с проверенным содержанием окиси кальция или по стандартному образцу известняка 59в. 0,1 г углекислого кальция растворяют в 100 мл соляной кислоты 1 :3 при нагревании, раствор кипятят 3—5 мин для удаления углекислоты, переводят в мерную колбу вместимостью 500 мл, охлаждают, разбавляют водой до метки и хорошо перемешивают.
Для установки титра раствора трилона Б на приборе ФЭТ-УНИИЗ отбирают пипеткой по 50 мл приготовленного раствора хлористого кальция в три стакана вместимостью по 150 мл, разбавляют водой до 80 мл, приливают из бюретки по 10—15 мл устанавливаемого раствора трилона Б, 15 мл раствора гидрата окиси калия, добавляют по 7 капель индикатора (хром темно-синего), опускают осторожно в стакан магнитный элемент, запаянный в стеклянную трубку, помещают стакан в гнездо прибора, включают прибор и мотор электромагнитной мешалки в соответствии с инструкцией; приложенной к прибору, переводят стрелку микроамперметра в крайнее правое положение шкалы, раствор перемешивают 1 мин и приступают к титрованию. Из бюретки по каплям приливают трилон Б до остановки стрелки микроамперметра, что соответствует эквивалентной точке.
Титр раствора трилона Б, выраженный в граммах СаО (7сао)» вычисляют по среднеарифметическому результату трех титрований по формуле
„ 50 .0,1 . %СаО
СаО V • 509 • 100 ’
где %СаО — содержание СаО в углекислом кальции, %;
V — объем 0,01 м раствора трилона Б, пошедший на титрование, мл.
При установке титра раствора трилона Б на приборе ТФЛ-46—2 перед началом титрования производят настройку прибора, которая заключается в следующем: тумблером «прибор 84 включен» включают титратор и в течение 20—30 мин дают ему прогреться, оптический клин устанавливают в нулевое положение, ручку чувствительности «грубо» — в минимальное крайнее левое положение. Бюретку с автоматическим нулем заполняют 0,01 м раствором трилона Б, стакан с раствором, содержащий кальций и все необходимые для его титрования реактивы, помещают в ячейку титрования, стрелку амперметра устанавливают на нулевую отметку, а ручку «чувствительности плавно» — на минимальную чувствительность (в крайнее левое положение). Затем в ячейку помещают предварительно оттитрованный обычным способом раствор вместо неоттитрованного, после чего поворачивают ручку «чувствительность плавно» по часовой стрелке до тех пор, пока не сработает система «конец титрования», что сигнализируется загоранием сигнальной лампы, после чего ручку «чувствительность плавно» поворачивают еще на 1—2 деления вправо с тем, чтобы автоматическое прекращение подачи титранта наступило несколько раньше эквивалентной точки. Стрелка амперметра при этом отклоняется на 20—25 делений. В том случае, если система не сработает, ручку «чувствительность грубо» поворачивают вправо на одно деление и повторяют операцию с «чувствительностью плавно» до тех пор, пока не произойдет перекрытие магистрали клапаном. После этого производят титрование анализируемого раствора с красным светофильтром 7. Скорость истечения титранта и скорость перемешивания раствора подбирается в процессе титрования. Нельзя допускать быстрого перемешивания, при котором образуется глубокая воронка в жидкости, что может привести к получению неправильного результата титрования.
Стакан, содержащий раствор кальция и все необходимые реактивы (щелочь, индикатор и др.), помещают в ячейку для титрования, включают магнитную мешалку и тумблер «автоматическое титрование», стрелку амперметра устанавливают на нуль ручкой «настройка нуля». Открывают кран бюретки и производят титрование. В конце титрования стрелка быстро отклоняется влево примерно на 20—25 делений, при этом подача титранта автоматически прекращается и загорается лампа «конец титрования». Установление конечной точки титрования осуществляется вручную следующим образом. Тумблер «двойное титрование» устанавливают в положение «включено», автоматическое титрование переключается на «визуальное», а стрелка амперметра осторожно переводится вправо на деление 30—40. Открывают кран бюретки и нажатием кнопки «подача титранта» производят титрование до полной остановки стрелки амперметра.
Титр раствора трилона Б, выраженный в граммах СаО (7сао), вычисляют по среднеарифметическому результату трех титрований по формуле в п. 3.6.
Фотоэлектрический титриметр ФЭТ-УНИИЗ и полуавтоматический титратор ТФЛ-46—2.
3.6.2. Проведение анализа
Из мерной колбы вместимостью 500 мл отбирают пипеткой 50 мл анализируемого раствора, полученного, как указано в п. 3.1, переносят его в стакан вместимостью 150 мл, добавляют 10 мл маскирующего реагента, 15 мл раствора щелочи, 7 капель индикатора хром темно-синего, опускают осторожно в стакан магнитный элемент, помещают стакан в гнездо прибора, включают прибор и мотор мешалки, как при установке титра, и титруют 0,01 м раствором трилона Б до остановки стрелки микроамперметра на ФЭТ-УНИИЗ или на ТФЛ-46—2 с визуальным окончанием.
Содержание окиси кальция (СаО) в процентах вычисляют по формуле
СаО =
V-TCaO -10. 100
м
где V — объем 0,01 м раствора трилона Б, пошедший на титрование, в мл;
Т'сао — титр 0,01 м раствора трилона Б, выраженный в г СаО; М — навеска цемента в г;
10 — коэффициент, учитывающий определение содержания СаО в аликвотной части фильтрата.
3.7. Определение содержания окиси магния
Принцип метода. Колориметрическое определение содержания окиси магния основано на образовании в щелочной среде гидроокиси магния, которая реагирует с красителем титановым желтым с образованием лака красного цвета. При этом краситель титановый желтый адсорбируется образующейся гидроокисью магния и дает прозрачные коллоидные растворы; для их устойчивости применяют защитный коллоид-крахмал и гидроксиламин. Железо и алюминий мешают определению магния, ослабляя окраску лака, поэтому их маскируют.
3.7.1. Применяемые реактивы, растворы и аппаратура
0,02%-ный раствор красителя титанового желтого; готовят следующим образом: 0,2 г красителя растворяют в 100 мл воды, раствор фильтруют в мерную колбу вместимостью 1 л. Отдельно растворяют 10 г гидроксиламина солянокислого по ГОСТ 5456—79 в 100 мл воды, нейтрализуют его 10%-ным раствором NaOH по наружному индикатору — фенолфталеиновой бумажке, фильтруют в ту же мерную колбу и раствор разбавляют водой до метки. Раствор готовят за сутки до применения и им можно пользоваться в течение месяца.
Маскирующий реагент; приготовление по п. 3.5.3.
Натрия гидрат окиси по ГОСТ 4328—77, 10%-ный раствор.
Крахмал растворимый (амилодекстрин) по ГОСТ 10163—76, 0,5%-ный водный раствор, свежеприготовленный.
Метиловый красный по ГОСТ 5853—51, 0,2% -ный спиртовой
раствор; готовят по п. 2.3.
Фотоэлектроколориметр.
Построение градуировочного графика. В три мерные колбы вместимостью 100 мл последовательно приливают 10; 20 и 50 мл стандартного раствора, приготовленного по п. 3.1.
Аликвотные части 10 и 20 мл разбавляют водой примерно до 50 мл, затем добавляют в колбы по 50 мл маскирующего реагента, по 5 мл крахмала, по 10 мл красителя титанового желтого и одну каплю индикатора метилового красного. Растворы нейтрализуют раствором едкого натра до лимонно-желтого цвета и добавляют избыток щелочи в 10 мл, разбавляют до метки водой, перемешивают, выдерживают 5 мин и колориметрируют в кювете размером 20 мм, пользуясь зеленым светофильтром (л=530—536 нм). По полученным значениям оптической плотности и известной концентрации эталонных растворов строят градуировочный график.
3.7.2. Проведение анализа
В две мерные колбы вместимостью 100 мл отбирают пипеткой по 20 мл анализируемого и стандартного растворов, полученных, как указано в п. 3.1, разбавляют водой примерно до 50 мл, добавляют маскирующий реагент, крахмал, краситель и производят все дальнейшие операции, вплоть до колориметрирования, как это указано при построении градуировочного графика.
Содержание окиси магния (MgO) в процентах вычисляют по формуле
100
М. V1 • 1000’
где Mi — количество окиси магния, найденное по графику, мг;
V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл; М — навеска цемента, г.
Примечания:
1. При наличии в цементе более 4—5% окиси магния его содержание может быть определено титрованием 0,05 м раствором трилона Б аликвотной части раствора 100 мл после отделения полуторных окислов, как это изложено в п. 2.7.
2. При выполнении контрольных анализов результаты определения содержания окиси магния необходимо проверить трилонометрическнм методом.
3.8. Определение содержания фосфорного ангидрида
А. С отделением кремнекислоты
Принцип метода. Колориметрический метод определения фосфора основан на образовании окрашенного в желтый цвет фосфор-нованадиевомолибденового комплексного соединения в присутствии азотной кислоты. При содержании 5—8 мл азотной кислоты в 100 мл раствора интенсивность окраски комплекса максимальная. Кремнекислота мешает определению фосфора, поэтому ее необходимо предварительно выделить или отогнать с фтористоводородной и азотной кислотами.
3.8.1. Применяемые реактивы, растворы и аппаратура
Кислота фтористоводородная по ГОСТ 10434—82, 40%-ный раствор.
Кислота азотная по ГОСТ 4461—77, плотностью 1,4 и ее растворы 1 :3 и 6 н.
Плавень; готовят по п. 2.9.
Стандартный раствор соли фосфата; готовят следующим образом: 0,1917 г дважды перекристаллизованного калия фосфорнокислого однозамещенного по ГОСТ 4198—75 растворяют в небольшом количестве воды, переносят в мерную колбу вместимостью 1 л, разбавляют до метки водой и хорошо перемешивают; 1 мл стандартного раствора содержит 0,1 мг P2Os.
Аммоний молибденовокислый по ГОСТ 3765—78, 5%-ный раствор.
Аммоний ванадиевокислый мета по ГОСТ 9336—75; готовят следующим образом: 2,5 г ванадиевокислого аммония растворяют в 500 мл горячей воды, охлаждают до комнатной температуры, добавляют 20 мл 6 н раствора азотной кислоты, разбавляют водой до 1 л и тщательно перемешивают.
Построение градуировочного графика. График строят по данным оптической плотности, полученным при колориметрировании серии эталонных растворов, приготовленных из стандартного раствора соли фосфата.
В четыре мерные колбы вместимостью 100 мл последовательно приливают 2; 4; 6; 8; 10 и 12 мл стандартного раствора фосфата с содержанием 0,1 мг Р2О5 в 1 мл, добавляют в каждую колбу по 5 мл азотной кислоты, разбавляют примерно до 50 мл водой, прибавляют по 10 мл раствора ванадиевокислого аммония, 10 мл молибдата аммония, доливают до метки водой, тщательно перемешивают и через 12—15 мин колориметрируют в кюветах размером 30 мм с синим светофильтром (1=400—420 нм).
Колориметрирование и построение градуировочного графика производят аналогично указанному в п. 3.1.
3.8.2. Проведение анализа
Навеску 1 г цемента смачивают в платиновом тигле несколькими каплями воды, прибавляют 4—5 капель азотной кислоты и 10—12 мл плавиковой кислоты. Содержимое тигля осторожно перемешивают платиновым шпателем и, не вынимая его, переносят тигель на нераскаленную песочную баню, отгоняют S!F4 и избыток HF при периодическом перемешивании. Сухой остаток сплавляют с небольшим количеством плавня и после охлаждения расплава выщелачивают его 30—40 мл горячей воды с добавлением 5—7 мл азотной кислоты. Раствор переводят в мерную колбу вместимостью 100 мл, охлаждают, доливают до метки водой и хорошо перемешивают. Для определения Р2О5 отбирают аликвотные части от 10 до 50 мл раствора в зависимости от предполагаемого содержания фосфорного ангидрида в цементе, помещают в мерную колбу вместимостью 100 мл, добавляют 6—3 мл азотной кислоты, разбавляют до 50 мл водой, прибавляют 10 мл раствора ванадиевокислого аммония, 10 мл молибдата аммония, доливают до метки водой, хорошо перемешивают и через 12—15 мин колориметрируют в кюветах размером 30 мм с синим светофильтром, как указано в п. 3.1.
Содержание фосфорного ангидрида (Р2О5) в процентах вычисляют по формуле
Mi - V * 100 м. vr • 1001
1000
где Л11 — количество Р2О5, найденное по графику, мг;
V — общий объем раствора в мл;
Vf — объем раствора, взятый для колориметрирования, в мл; М — навеска цемента в г.
Б. Без отделения кремнекислоты
Принцип метода. Колориметрический метод определения фосфора основан на образовании окрашенного в синий цвет комплекса/фосфорномолибденовой гетерополикислоты молибдатом натрия при кислотности раствора, примерно 1 н по серной кислоте или с молибдатом аммония при кислотности раствора 0,5—0,6 н по соляной кислоте, восстановленного соответственно сернокислым гидразином или метолом.
В указанных выше условиях кислотности раствора кремнемолибденовая гетерополикислота не образуется. Определение проводят соответственно из аликвотной части основного солянокислого раствора, приготовленного, как указано в п. 3.1, или из отдельной навески.
3.8.3. Применяемые реактивы, растворы и аппаратура
Стандартный раствор соли фосфата; готовят следующим образом: 100 мл стандартного раствора, приготовленного, как указано в п. 3.8А, разбавляют до 500 мл; 1 мл приготовленного раствора содержит 0,02 мг Р2О5.
Плавень; готовят по п. 2.9.
Натрий молибденовокислый по ГОСТ 10931—74, 2,5%-ный
раствор в 10 н серной кислоте; готовят следующим образом: 5 г молибденовокислого натрия растворяют в 200 мл 10 н раствора серной кислоты.
Восстановитель 1; гидразин сернокислый по ГОСТ 5841—74. 0,15%-ный водный раствор.
Калия гидрат окиси, 3 н раствор.
Кислота соляная по ГОСТ 3118—77, раствор 1 :3.
Восстановитель 2; готовят следующим образом: 20 г метола (пара-метиламиносульфата) по ГОСТ 25664—83, 12 г сернистокислого натрия (натрия сульфита) безводного по ГОСТ 195—77 растворяют в 500 мл теплой воды, отфильтровывают и разбавляют водой до 1 л. Хранят в склянке темного стекла.
Аммоний молибденовокислый по ГОСТ 3765—78, 5%-ный раствор; готовят по п. 3.1.
Кислота лимонная по ГОСТ 3652—69, 4%-ный раствор. Фотоэлектроколориметр.
Построение градуировочного графика с восстановителем 1. График строят по данным оптической плотности, полученным при колориметрировании серии эталонных растворов, приготовленных из стандартного раствора соли фосфата. В четыре мерные колбы вместимостью 50 мл последовательно приливают 1; 2; 3 и 4 мл стандартного раствора фосфата с содержанием 0,02 мг Р2О5 в 1 мл, добавляют в каждую колбу воды до 25—30 мл общего объема, по 5 мл раствора молибдата натрия, по 2 мл сернокислого гидразина и доливают до метки водой. Растворы хорошо перемешивают и погружают на 10 мин в кипящую водяную баню, после чего их охлаждают до комнатной температуры и колориметрируют в кювете размером 20 мм с красным светофильтром (Х=656—680 нм).
Построение градуировочного графика производят, как указано в п. 3.1.
3.8.4. Проведение анализа
Отбирают пипеткой 25 мл анализируемого раствора (первая порция), полученного, как указано в п. 3.1, переносят его в мерную колбу вместимостью 50 мл. Предварительно 25 мл этого же раствора (вторая порция), отобранного в стакан или колбу, нейтрализуют 3 н раствором гидроокиси калия по фенолфталеину. Пошедшее на нейтрализацию 25 мл (вторая порция), количество щелочи прибавляют к отобранной аликвотной части (первая порция), затем добавляют 5 мл раствора молибдата натрия, 2 мл раствора сернокислого гидразина, доливают до метки водой, перемешивают и после 10 мин выдерживания в кипящей водяной бане, охлаждают и колориметрируют. Содержание фосфорного ангидрида (Р2О5) в процентах вычисляют по формуле, приведенной в п. 3.8.2.
Построение градуировочного графика с восстановителем 2. График строят по известной концентрации и оптической плотности, полученной в результате колориметрирования серии эталонных растворов, приготовленных из стандартного раствора соли фосфата. В четыре мерные колбы вместимостью 100 мл последовательно приливают 5; 10; 15 и 25 мл стандартного раствора, добавляют в каждую колбу воды до 25 мл общего объема, по 20 мл раствора соляной кислоты, 20 мл раствора восстановителя и 5 мл раствора молибдата аммония, перемешивают и дают постоять 10 мин для полного образования фосфорномолибденового комплекса. Затем добавляют по 10 мл раствора лимонной кислоты, разбавляют водой до метки и хорошо перемешивают. Колориметрируют полученные синие растворы и строят градуировочный график.
3.8.5. Проведение анализа
Навеску 0,5 г цемента сплавляют с 2 г плавня. После охлаждения плав выщелачивают в 50 мл раствора соляной кислоты и переводят в мерную колбу вместимостью 250 мл, разбавляют водой до метки и хорошо перемешивают. Для определения содержания фосфорного ангидрида отбирают аликвотную часть раствора в 50 мл, помещают в мерную колбу вместимостью 100 мл, добавляют 10 мл раствора соляной кислоты, а также все остальные реактивы, указанные в п. 3.8.4, при описании построения градуировочного графика. Колориметрируют полученный раствор и вычисляют содержание фосфорного ангидрида, как указано в п. 3.8.2.
3.9. Определение содержания окиси хрома
Принцип метода. Колориметрический метод основан на окислении в кислой среде днфенилкарбазида шестивалентным хромом с образованием растворимого соединения, окрашенного в краснофиолетовый цвет. Окраска соединения максимальна и устойчива при кислотности раствора 0,20—0,25 н по серной кислоте; реакция протекает в течение нескольких секунд. Мешающее влияние трехвалентного железа устраняют путем переведения его в бесцветный фосфатный комплекс.
3.9.1. Применяемые реактивы, растворы и аппаратура
Плавень; готовят по п. 2.9.
Кислота соляная по ГОСТ 3118—77, плотностью 1,19 и ее раствор 1 :3.
Кислота ортофосфорная по ГОСТ 6552—80, раствор 1 :2.
Дифенилкарбазид по ГОСТ 5859—78, 0,25% -ный раствор, готовят следующим образом: навеску 0,25 г днфенилкарбазида растворяют при слабом нагревании в 100 мл эталового ректификованного спирта по ГОСТ 5962—67, разбавленного водой 1:1. Дифенилкарбазид нестоек и его раствор готовят перед определением хрома.
Стандартный раствор 1; готовят на основе стандартного образца хромомагнезитового кирпича № 239 с содержанием окиси хрома 22,02%. Стандартный образец в количестве 0,05 г сплавляют с 1 г плавня, плав выщелачивают 40—50 мл раствора соляной кислоты без подогревания, во избежание восстановления хрома до трехвалентного, переводят в мерную колбу вместимостью 500 мл, разбавляют до метки водой и хорошо перемешивают. 1 мл стандартного раствора содержит 0,022 мг окиси хрома.
Стандартный раствор 2; готовят следующим образом: 0,0484 г очищенного перекристаллизацией и высушенного при температуре 130° С калия двухромовокислого по ГОСТ 4220—75 растворяют в 100 мл воды, переводят в мерную колбу вместимостью 250 мл, разбавляют водой до метки и тщательно перемешивают. Аликвотную часть этого раствора 50 мл разбавляют водой до 250 мл. 1 мл стандартного раствора содержит 0,02 мг окиси хрома.
Фотоэлектроколориметр.
Построение градуировочного графика. Для приготовления эталонных растворов в пять мерных колб вместимостью 100 мл последовательно приливают 0,4; 0,8; 1,2; 1,6 и 2 мл стандартного раствора, разбавляют примерно до 70 мл водой, добавляют в каждую колбу по 1,5 мл соляной кислоты, 4 мл фосфорной кислоты, после чего хорошо перемешивают, прибавляют по 1,5 мл дифенил-карбазида, разбавляют водой до метки, снова хорошо перемешивают и через 5—7 мин колориметрируют в кювете размером 30 мм с зеленым светофильтром (л=530—536 нм). По полученным величинам оптических плотностей и известным концентрациям эталонных растворов строят градуировочный график, как указано в п. 3.1.
3.9.2. Проведение анализа
Навеску 0,2—0,5 г цемента сплавляют с 2 г плавня. После охлаждения сплав выщелачивают 40—50 мл раствора соляной кислоты, переводят в мерную колбу вместимостью 100 мл, разбавляют до метки водой и хорошо перемешивают.
Для определения окиси хрома отбирают аликвотные части от 10 до 25 мл раствора в зависимости от предполагаемого содержания окиси хрома в цементе, помещают в мерную колбу вместимостью 100 мл, добавляют 50—60 мл воды, 4 мл раствора фосфорной кислоты и хорошо перемешивают. После чего прибавляют 1,5 мл раствора дифенилкарбазида, разбавляют водой до метки, снова хорошо перемешивают и через 5—7 мин производят колориметрирование.
Содержание окиси хрома (СггО3) в процентах вычисляют по формуле
где Mi — количество Сг20з, найденное по графику, мг;
V — общий объем раствора, мл;
Vi — объем раствора, взятый для колориметрирования, мл; М—навеска цемента, г.
3.10. Определение содержания фтора
Принцип метода. Колориметрический метод определения содержания фтора основан на ослаблении интенсивности окраски комплексного соединения алюминия с арсеназо 1 в присутствии ионов фтора, с которым алюминий образует более устойчивый комплекс AlFe3”. Комплекс ионов алюминия с арсеназо 1 имеет наиболее интенсивную и устойчивую окраску при величине pH 4,5. Ослабление окраски арсеназо-алюминиевого комплекса очень чувствительно к величине pH раствора и зависит от солевого фона фотомет-рируемого раствора, поэтому при измерении оптических плотностей растворов pH должно точно равняться 4,5 и необходимо, чтобы измеряемые растворы имели одинаковый солевой состав.
3.10.1. Применяемые реактивы, растворы и аппаратура
Кислота соляная по ГОСТ 3118—77, растворы 0,1 ни 1:3.
Аммиак водный по ГОСТ 3760—79, 25%-ный, 10%-ный и
0,2%-ный растворы.
Натрий углекислый безводный по ГОСТ 83—79 или калий-натрий углекислый безводный по ГОСТ 4332—76.
Плавень; готовят следующим образом: 1,7 весовые части лития углекислого по МРТУ 6—09—4806—67 и 1,3 весовые части борной кислоты по ГОСТ 9656—75 тщательно смешивают в фарфоровой ступке. Полученную смесь высушивают в сушильном шкафу при температуре 160—200° С в течение 2 ч, затем растирают в фарфоровой ступке и добавляют 1 весовую часть окислителя — аммония азотнокислого по ГОСТ 22867—77. Если такая смесь при сплавлении вспучивается или разбрызгивается, то ее следует еще раз высушить и при растирании снова добавить окислитель.
Арсеназо 1 по МРТУ 6—09—3546—67; 0,0525%-ный раствор;
готовят следующим образом: 0,2625 г арсеназо 1 растворяют в 300 мл воды, переводят в мерную колбу вместимостью 500 мл, доводят до метки водой и хорошо перемешивают.
Стандартный раствор фторида натрия; готовят следующим образом: 0,2212 г очищенного перекристаллизацией и высушенного при температуре 120° С натрия фтористого по ГОСТ 4463—76 растворяют в 200—300 мл воды, переводят в мерную колбу вместимостью 1 л и тщательно перемешивают. Аликвотную часть этого раствора 100 мл разбавляют до 1 л; 1 мл стандартного раствора содержит 0,01 мг фтора.
Алюминий азотнокислый по ГОСТ 3757—75, 0,001 м раствор; готовят следующим образом: 0,0938 г А1 (МО3)з-9Н2О растворяют в 100—150 мл воды, переводят в мерную колбу вместимостью 250 мл и хорошо перемешивают. Раствор соли алюминия рекомендуется хранить не более 15—20 дней.
Ацетатно-аммиачный буферный раствор; готовят следующим образом: 8,7 мл 25%-ного аммиака водного по ГОСТ 3760—79 помещают в мерную колбу вместимостью 1 л, добавляют 200 мл воды, 15 мл 80%-ной или 11 мл ледяной уксусной кислоты по ГОСТ 61—75, 700—750 мл воды и, не доводя до метки, перемешивают и определяют на потенциометре или pH-метре величину pH. При отклонении pH на 0,05 корректируют приготовленный раствор до значения pH 4,5 добавлением аммиака или ледяной уксусной кислоты.
Холостой раствор; готовят на основе сырьевого шлама, не содержащего фтора. Навеску стандартного шлама 0,5 г смешивают с 1 г углекислого натрия или калия-натрия углекислого, взвешенных с точностью ±0,01 г (для соблюдения постоянства солевого фона раствора) и спекают или сплавляют с 1 г плавня при температуре не выше 900—950°С в течение 3—5 мин. После охлаждения спек растворяют в 50 мл раствора соляной кислоты 1 :3, нагретого до 50—60е С. Борно-литиевый сплав растворяют в 50 мл раствора соляной кислоты 1 :3 на магнитной мешалке при нагревании в течение 20 мин, не доводя до кипения.
Полученные растворы нагревают до 80—90° С, осаждают алюминий и железо 10%-ным раствором аммиака в присутствии одной капли индикатора метилового красного до перехода окраски раствора в желтый цвет и дают избыток аммиака 2—3 капли. Горячий раствор фильтруют через неплотный фильтр, промывают 3—4 раза горячим 0,2%-ным раствором аммиака.
Фильтрат и промывные воды собирают в мерную колбу вместимостью 250 мл, доводят до метки водой и хорошо перемешивают.
Метиловый красный по ГОСТ 5853—51, 0,2%-ный спиртовой
раствор; готовят по п. 2.3.
Электромагнитная мешалка.
Фотоэлектроколориметр.
рН-метр.
Построение градуировочного графика. В 4 мерные колбы вместимостью 50 мл последовательно приливают по 5 мл холостого раствора, а затем во все колбы, кроме первой (нулевой раствор), добавляют соответственно по 0; 1; 2 и 3 мл стандартного раствора и по 2 мл раствора арсеназо 1. Окраска реагента имеет различные оттенки в зависимости от величины pH раствора, в который его добавляют (в щелочной среде арсеназо 1 имеет малиновую окраску, а при pH 4,5 — оранжевую). Полученные растворы нейтрализуют 0,1 н раствором соляной кислоты до перехода окраски раствора из малиновой в красно-оранжевую (более в оранжевую). Затем в каждую колбу вводят по 2 мл азотнокислого алюминия, по 10 мл буферного раствора, доводят до метки дистиллированной водой, хорошо перемешивают и дают постоять 15 мин. Оптическую плотность измеряют на фотоэлектроколориметре с желтым светофильтром (Х=582 нм) или с зеленым светофильтром (Х=530—536 нм) в кюветах толщиной слоя 20 мм. Раствор первой колбы используют в качестве нулевого раствора сравнения.
Измерение оптической плотности производят следующим образом: в две кюветы наливают нулевой раствор и помещают их в левый и правый кюветодержатели на пути светового пучка, в третью кювету наливают раствор, содержащий фтор. Оптическую плотность первого барабана устанавливают на величину 0,350, а стрелку гальванометра переводят на нуль. Относительно плотности нулевого раствора замеряют оптическую плотность всех остальных растворов с различным содержанием фтора. При этом оптическая плотность растворов, содержащих фтор, будет ниже 0,350 и с увеличением его концентрации будет соответственно уменьшаться. На основании полученных оптических плотностей и известной концентрации фтора строят градуировочный график, как это изложено в п. 3.1.
3.10.2. Проведение анализа
Навеску цемента 0,1—0,5 г в зависимости от содержания фтора спекают с 1 г углекислого натрия или калия-натрия углекислого или сплавляют с 1 г борно-литиевого плавня при температуре 900—950° С в течение 3—5 мин. Полученные спек и сплав выщелачивают в растворе соляной кислоты 1 :3 так же, как при приготовлении холостого раствора, отделяют полуторные окислы и переводят фильтраты в мерную колбу вместимостью 250 мл, доводят водой до метки и хорошо перемешивают. Аликвотную часть анализируемого раствора 1—5 мл помещают в мерную колбу вместимостью 50 мл. Для соблюдения постоянства солевого состава в колбу добавляют холостой раствор в количестве 4—0 мл в зависимости от величины аликвотной части анализируемого раствора (общий объем анализируемого и холостого растворов должен составлять 5 мл). В другую мерную колбу вместимостью 50 мл вводят 5 мл холостого раствора, затем в обе колбы добавляют по 2 мл арсеназо 1, 0,1 н раствора соляной кислоты до перехода
окраски раствора в красно-оранжевую, по 2 мл азотнокислого алюминия, по 10 мл буферного раствора и доводят до метки водой. Остальные операции производят так, как описано при построении графика в п. 3.1. В качестве нулевого раствора служит колба с холостым раствором.
Содержание фтора (F) в процентах вычисляют по формуле
р== Лу У - 100 м • Vj • 1000’
где Л11 — количество фтора, найденное по графику, мг;
V — общий объем раствора, мл;
Vt — объем раствора, взятый для колориметрирования, мл;
М — навеска цемента, г.
3.11. Определение содержания хлора
Принцип метода. Нефелометрический метод определения содержания хлора основан на увеличении оптической плотности раствора вследствие образования суспензии ионами хлора с ионами серебра в присутствии азотной кислоты. Переведение образца в растворимое состояние осуществляют сплавлением с низкотемпературной смесью буры с калий-натрий углекислым, во избежание улетучивания хлора.
3.11.1. Применяемые реактивы, растворы и аппаратура
Стандартный раствор соли хлора; готовят следующим образом: навеску 0,0165 г предварительно дважды перекристаллизованного и высушенного при температуре 110° С натрия хлористого по ГОСТ 4233—77 растворяют в 100 мл воды, переносят в мерную колбу вместимостью 250 мл, разбавляют до метки водой и хорошо перемешивают. 1 мл стандартного раствора содержит 0,004 мг хлора.
Кислота азотная по ГОСТ 4461—77, раствор 1 : 5.
Серебро азотнокислое по ГОСТ 1277—75, 5%-ный раствор.
Смесь для сплавления; готовят следующим образом: 1 весовая часть натрия тетраборнокислого безводного по ГОСТ 4199—76 и 2 весовые части калия-натрия углекислого безводного по ГОСТ 4332—76 тщательно смешивают в фарфоровой ступке, добавляя небольшими порциями одно к другому.
Холостой раствор; готовят следующим образом: навеску 0,5 г сырьевой смеси, не содержащей хлора, перемешивают с 2 г плавня, сплавляют в муфеле при температуре 700—720° С в течение 15 мин. По охлаждении сплав растворяют в 40 мл холодного раствора азотной кислоты на магнитной мешалке. Прозрачный раствор переводят в мерную колбу вместимостью 250 мл, доводят водой до метки и перемешивают.
Магнитная мешалка.
Фотоэлектроколориметр.
Построение градуировочного графика. График строят по данным оптической плотности, полученным при колориметрировании серии эталонных растворов, приготовленных из стандартного раствора соли хлора.
В мерные колбы вместимостью 100 мл приливают по 50 мл холодного раствора и последовательно добавляют 1; 3 и 5 мл стандартного раствора соли хлора. Колбы с растворами помещают в водяную баню с температурой 80—90° С и выдерживают в ней до удаления пузырьков углекислого газа, но не более 5 мин; растворы охлаждают и добавляют по 2 мл азотнокислого серебра, доливают до метки водой, тщательно перемешивают 1 мин и 15 мин выдерживают в темном месте, после чего нефелометрируют в кюветах размером 50 мм с синим светофильтром (Х=420—453 нм), как это изложено в п. 3.1.
3.11.2. Проведение анализа
Навеску 0,5 г цемента сплавляют с 2 г плавня в течение 15 мин при температуре 700—720° С. Получают раствор анализируемого цемента, как при приготовлении холостого раствора, отбирают аликвотную часть раствора в количестве 50 мл и далее поступают, как это описано при построении градуировочного графика.
Содержание хлора (С1) в процентах вычисляют по формуле с1= V < * ИЮ
~ . М • 1000’
где V — общий объем раствора, мл;
Vt — объем раствора, взятый для нефелометрирования, мл;
М — навеска цемента, г;
-Mi — количество хлора, найденное по графику, мг.
СОДЕРЖАНИЕ
3. Методы испытаний материалов
ГОСТ 310.1—76 Цементы. Методы испытаний. Общие положения ... 3
(СТ СЭВ 3920—82)
ГОСТ 310.2—76 Цементы. Методы определения тонкости помола . 5
(СТ СЭВ 3920—82)
ГОСТ 310.3—76 Цементы. Методы определения нормальной густоты, (СТ СЭВ 3920—82)
сроков схватывания и равномерности изменения объема О ГОСТ 310.4—81 Цементы. Методы определения предела прочности при (СТ СЭВ 3920—82)
изгибе и сжатии
ГОСТ 310.5—80 Цементы. Методы определения теплоты гидратации .
ГОСТ 5382—73 Цементы. Методы химического анализа
ГОСТ 5802—78 Растворы строительные. Методы испытаний ....
ГОСТ 8735—75 Песок для строительных работ. Методы испытаний . .
ГОСТ 8269—76 Щебень из естественного камня, гравий и щебень из
вия для строительных работ. Методы испытаний .
ГОСТ 9552—76 Цементы. Глиноземистый, высокоглиноземистый и гипсоглиноземистый расширяющийся. Методы химического анализа
ГОСТ 9758—77 Заполнители пористые неорганические для бетона. Ме
тоды испытаний
ГОСТ 22236—76 Цементы. Правила приемки
ГОСТ 22237—76 Цементы. Упаковка, маркировка, транспортирование и
хранение
ГОСТ 25094—82 Добавки активные минеральные. Методы испытаний .
ГОСТ 25589—83 Щебень, гравий и песок для строительных работ. Методы
определения содержания сернокислых и сернистых соединений
БЕТОН И ЖЕЛЕЗОБЕТОННЫЕ ИЗДЕЛИЯ Часть 2
Редактор Р. Г. Говердовская Технический редактор Н. В. Келейникова Корректор М, С. Кабаиюва
Сдано в набор 29.06.84. Подп. к печати 14.03.85. Формат 60Х90’Лв. Бумага типографская № 2.
Литературная гарнитура. Высокая печать. 18,0 усл. п. л. 18,13 усл. кр.-отт. 19.17 уч.-нзд. л.
Ордена «Знак Почета» Издательство стандартов, 123840, Москва ГСП,
Новопресненский пер., д. 3.
Великолукская городская типография управления издательств,
полиграфии и книжной торговли Псковского облисполкома.
182100, г. Великие Луки. ул. Полиграфистов, 78/12
